Comments (4)
 cfrioux
commented on May 27, 2024
cfrioux
commented on May 27, 2024
Hi @KDeaton
- If you are mentioning the genomes collection of 1,520 culturable species from the human gut microbiota, then yes, we ran the workflow separately for each set of targets. But you could run the pipeline for a wide list of targets without separating them, there are no technical restrictions.
- Do you have an example of such warnings?
- I'll let @ArnaudBelcour answer that third question. A brief and incomplete answer would be to separate the recon step from the rest of the pipeline and directly use the underlying mpwt package.
from metage2metabo.
 ArnaudBelcour
commented on May 27, 2024
ArnaudBelcour
commented on May 27, 2024
Hi @KDeaton,
-
To be more precise on the targets used in the article for the 1,520 culturable species in the human gut: we used the addedvalue (i.e. the metabolites producible by the community but not by individual alone) as the targets for the
m2mpipeline (so for this pipeline we have only one list of targets). This allow us to find the key species associated to these targets.
Then we classified these metabolites in 6 different categories (such as lipids or sugar). And we used each of these 6 groups as targets for them2m_analysispipeline to visualize the minimal communities with powergraphs. But we used this because we had a lot of targets in the addedvalue (156 metabolites). -
As annotation quality do you refer to the genome annotation or the SBML quality?
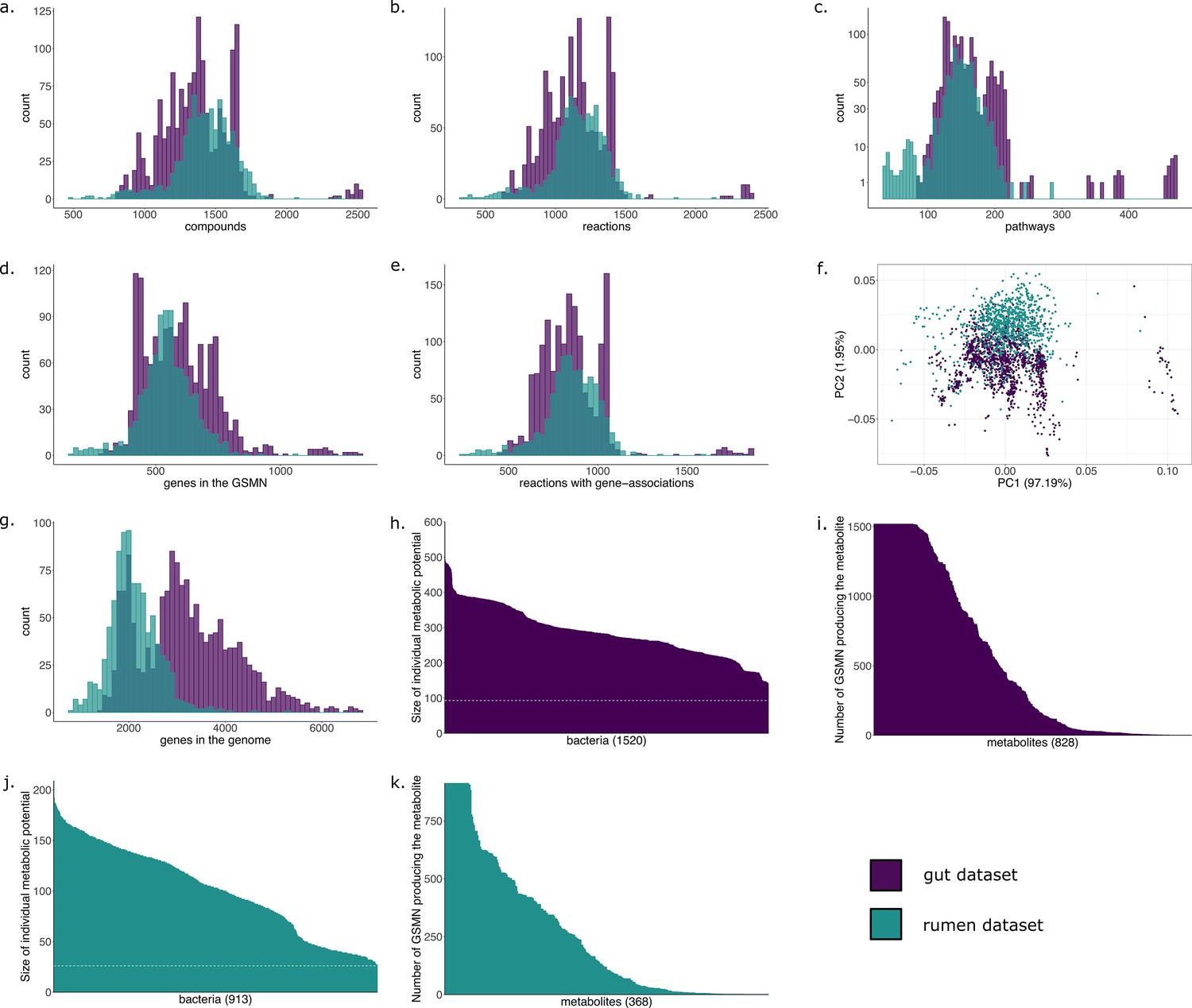
For the genome annotation, it is quite difficult to estimate the good quality of an annotation especially when dealing with metagenomics data. But it will depend on the tool used for the annotation such as Prokka or eggnog-mapper (Prokka being fast but less accurate than Eggnog-mapper). And there can big some big variations (for example in our article we have variations between genomes associated to 500 reactions to genomes associated to 2,500 reactionss, as you can see in the subfigure b. of this supplementary figure).
For the SBML quality, the SBML files created by metage2metabo contain few annotations. And for example, SBML quality check tools (such as memote) might put a very low score to those SBML. Nonetheless, the information they contain is sufficient for m2m. -
There is no command to ignore the failed builds with m2m. An easy option if you want to continue the analysis without the failed builds is to remove them from the input folder. You can find the failed build using the
resume_inference.tsvinside the folderm2m_output_folder/pgdb_log, the failed builds have anERRORin their gene_number column. By relaunching m2m, this will uses the successful builds stored in ptools-local folder and creates the corresponding SBML files.
Another way is to keep the failed builds and try to create the PGDB files for the successful builds. There is a possible work-around with mpwt. I have released a new version of mpwt recently (0.7.0) that refactor how mpwt works. With this version each run is independent so if one fails the other will still be process till their ends. So in this case it will produce the PGDB files for m2m.
If you can't update to this version, there is an option with older version of mpwt--ignore-errorthat will allow to continue the draft reconstruction even if some build have failed.
In both case, you have to use mpwt commandmpwt -f m2m_input_folder -o m2m_output_folder/pgdb --patho --flat --md -v --cpu Xand by adding--ignore-errorif you used the second option. But this will only produce PGDB files for the successful builds and it will not create the SBML files. To go further you need to fix the issue with the failed builds or remove them.
To find why some builds failed you can take a look at thepathologic.logfiles located in the input folder. They should contain the errors encountered by Pathway Tools during the inference.
{kind=link}
from metage2metabo.
 KDeaton
commented on May 27, 2024
KDeaton
commented on May 27, 2024
Thanks for both of your responses! I'm all set with questions 1 & 3.
For more information on my question 2, when I ran a large metagenome that had a few builds fail, the resume_inference.tsv listed at least 10 in the pwt_warning column. When I ran recon again on a subset of genomes that had successful builds, the process finishes successfully and creates the sbml files, though I didn't get a resume_inference.tsv file. When I check the pathologic.log, there are several warnings. Here are some examples:
Warning: The Location "join(1450558..1451127,1..24)" shows a first
basepair number that is bigger than the second. This should
only happen when crossing the origin.
Warning: tRNA IPF37_06710 (NIL) may not have had parsable anticodon
information. None assigned.
No reaction or class having EC number 5.6.2.c can be found in the MetaCyc DB.
Warning: enter-into-lookup-table-internal: Why does acylactivating have 53 associated reactions??
from metage2metabo.
ArnaudBelcour
commented on May 27, 2024
Thanks for the examples I better understand you question now.
These warnings come from Pathway Tools and they can have multiple meanings:
- a certain number of the warnings are only for informations. They will not have an impact on the metabolic network. For example, it will be about consistency between gene location and nucleic sequence (but this can vary according to the codon table used).
- other warning indicates that some specific informations are not found (such as the EC example which is a strange EC because a 'correct' EC has only number in it and not letter). This can come from the format of the information (for example wrong format for GO Terms or EC number). It can be the result of old version of tools used for the annotation of the genomes (which can use old version of the ontology associated to these informations). But it is possible to get it with current tool as standardization of informations is quite difficult.
- some warnings are here to inform the user that some data in its input files are incompatible with some step of Pathway Tools. For example, I have often this issue when working with some datasets containing only proteins. As I do not have nucleic sequence the Hole Filler option will not be working on these data.
- some warnings are associated to check on the metabolic network. The purpose of PathoLogic is to create draft metabolic network but as they are drafts they can require manual curation by the user to be sure that the associations are correct. I think your last warning is more in this case (I think the issue is the fact that an enzyme is associated to 53 reactions).
I put a print of these warnings but it is more an informations for the user. Some warnings can need a manual curation (to keep or not the reaction proposed/associated to the gene). For example, in your last example it can be interesting to look at the 53 reactions associated to an enzyme. The issue is when dealing with hundred/thousand of reconstructions we can not have the time to check all of them.
For the fact that mpwt did not produce log at your second run I will look into this to try to find why it failed.
from metage2metabo.
Related Issues (20)
- I have the following problems when running the t2d_m2m_target_producers.R file: HOT 8
- Multiple errors when running test data HOT 7
- A better focus on the host metabolism when a host is provided HOT 1
- Issue with pathway tools API limitation and PGDB entries verification HOT 4
- Compute deadend and orphan metabolites
- Non optimal powergraph visualisation HOT 2
- m2m_analysis powergraph not working as expected HOT 27
- m2m recon not working with gbff file inputs HOT 4
- Error with analysis graph when using taxon_id HOT 5
- CRITICAL:metage2metabo.m2m.reconstruction:Something went wrong running Pathway Tools. See the log file in /home/chencong/output3/pgdb_log/log_error.txt HOT 6
- No /home/chencong/.ncbirc file, please fix it before using the program HOT 5
- [Question] Where can I find seed files? HOT 4
- Install metage2metabo problem HOT 5
- How to prepare inputs for metage2metabo HOT 3
- mpwt could not find the version of Pathway Tools HOT 1
- Individual scopes include seeds even if non producible or absent from the GSMNs HOT 3
- Do not allow abbreviation in command arguments. HOT 1
- Sanitize use of tarfile extractall. HOT 1
- gff file format problem while running m2m recon HOT 1
- When using `--pwt-xml`, the xml files extracted from Pathway Tools can be incompatible with python-libsbml. HOT 1
Recommend Projects
-
 React
React
A declarative, efficient, and flexible JavaScript library for building user interfaces.
-
Vue.js
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
-
 Typescript
Typescript
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
-
TensorFlow
An Open Source Machine Learning Framework for Everyone
-
Django
The Web framework for perfectionists with deadlines.
-
 Laravel
Laravel
A PHP framework for web artisans
-
D3
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
-
Recommend Topics
-
javascript
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
-
web
Some thing interesting about web. New door for the world.
-
server
A server is a program made to process requests and deliver data to clients.
-
Machine learning
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
-
Visualization
Some thing interesting about visualization, use data art
-
Game
Some thing interesting about game, make everyone happy.
Recommend Org
-
Facebook
We are working to build community through open source technology. NB: members must have two-factor auth.
-
Microsoft
Open source projects and samples from Microsoft.
-
Google
Google ❤️ Open Source for everyone.
-
Alibaba
Alibaba Open Source for everyone
-
D3
Data-Driven Documents codes.
-
Tencent
China tencent open source team.
from metage2metabo.