Comments (10)
 alchemistcai
commented on May 28, 2024
1
alchemistcai
commented on May 28, 2024
1
- If you rename HIS to HID manually,do it after Reduce removing and adding Hs,not before.
- Reduce will generate HID and HIE both but won't change atom name.Renaming HIS to HID and using Reduce will cause wrong protonation.
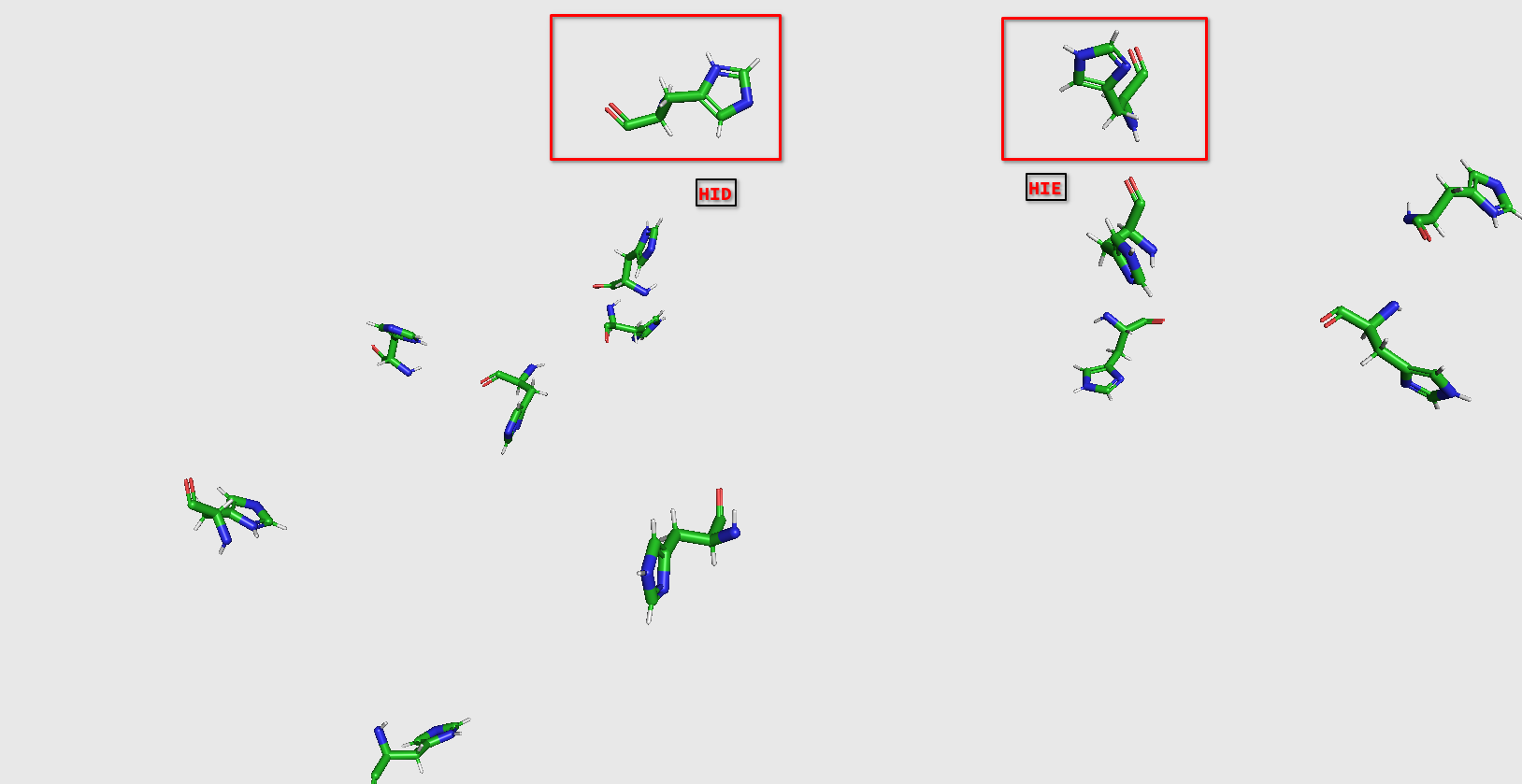
- I have a solution:if you have installed GROMACS,use
pdb2pqr30 --ff=AMBER --ffout=AMBER --with-ph=7.4 github_protein.pdb gpamber.pqr,and export it to pdb by Pymol.It will generate correct atom type. - Are there other more elegant solutions?Ambertools?I haven't figure it out.Maybe some PDB tools are more friendly on Amber FFs.
- This pdb file may be used for the simulation directly.Try to run with it.Good luck.
github_pdb_pqr.zip
from lightdock.
alchemistcai
commented on May 28, 2024
Upload your structure.pdb may help us know what's wrong.
from lightdock.
 brianjimenez
commented on May 28, 2024
brianjimenez
commented on May 28, 2024
Hi @mhyleung ,
You may need to rename HIS to HID in the protein partner. Then, you can follow these steps:
reduce -Trim prot.pdb > prot_no_h.pdb;
reduce -BUILD prot_no_h.pdb > prot_h.pdb;
reduce -Trim dna.pdb > dna_no_h.pdb;
reduce -BUILD dna_no_h.pdb > dna_h.pdb;
python reduce_to_amber.py dna_h.pdb fixed_dna_h.pdb;You can use this more updated version of the reduce_to_amber.py script:
#!/usr/bin/env python3
import os
import argparse
from lightdock.scoring.dna.data.amber import atoms_per_residue
from lightdock.pdbutil.PDBIO import read_atom_line
def _format_atom_name(atom_name):
"""Format ATOM name with correct padding"""
if len(atom_name) == 4:
return atom_name
else:
return " %s" % atom_name
def write_atom_line(atom, output):
"""Writes a PDB file format line to output."""
if atom.__class__.__name__ == "HetAtom":
atom_type = "HETATM"
else:
atom_type = "ATOM "
line = "%6s%5d %-4s%-1s%3s%2s%4d%1s %8.3f%8.3f%8.3f%6.2f%6.2f%12s\n" % (
atom_type,
atom.number,
_format_atom_name(atom.name),
atom.alternative,
atom.residue_name,
atom.chain_id,
atom.residue_number,
atom.residue_insertion,
atom.x,
atom.y,
atom.z,
atom.occupancy,
atom.b_factor,
atom.element,
)
output.write(line)
translation = {"H5'":"H5'1", "H5''":"H5'2", "H2'":"H2'1", "H2''":"H2'2", "OP1":"O1P", "OP2":"O2P"}
if __name__ == "__main__":
parser = argparse.ArgumentParser()
parser.add_argument("input_pdb_file")
parser.add_argument("output_pdb_file")
args = parser.parse_args()
with open(args.input_pdb_file) as ih:
with open(args.output_pdb_file, 'w') as oh:
for line in ih:
line = line.rstrip(os.linesep)
if line.startswith("ATOM "):
atom = read_atom_line(line)
if atom.residue_name not in atoms_per_residue:
print(f"[Warning] Not supported atom: {atom.residue_name}.{atom.name}")
else:
if atom.name not in atoms_per_residue[atom.residue_name]:
try:
atom.name = translation[atom.name]
write_atom_line(atom, oh)
except KeyError:
print(f"[Warning] Atom not found in mapping: {atom.residue_name}.{atom.name}")
else:
write_atom_line(atom, oh)If after this "protocol" you're still having issues with some atom types, it might be the capping at the C or N-terminal. Try to remove those hydrogens, remove all LightDock generated files in that directory and try again.
Let us know if this fixes your issues, otherwise you may follow @alchemistcai comment and share the structure (if possible).
from lightdock.
 mhyleung
commented on May 28, 2024
mhyleung
commented on May 28, 2024
Thank you for the rapid response. Neither the new script nor changing the HIS residues to HID helped. The error now simply is
[lightdock] ERROR: [NotSupportedInScoringError] Residue A.HID.282 or atom HE2 not supported. DNA scoring only supports AMBER94 types.
I have tried it also on another protein/DNA match constructed based on a similar fashion, run the new script and tried. The same HIS error occurred. This time, I noticed that when I change the HIS to HID, the reduce -BUILD step gives me this in the middle of the output:
*WARNING*: Res "HID" not in HETATM Connection Database. Hydrogens not added.
*WARNING*: Res "HID" not in HETATM Connection Database. Hydrogens not added.
*WARNING*: Res "HID" not in HETATM Connection Database. Hydrogens not added.
*WARNING*: Res "HID" not in HETATM Connection Database. Hydrogens not added.
*WARNING*: Res "HID" not in HETATM Connection Database. Hydrogens not added.
*WARNING*: Res "HID" not in HETATM Connection Database. Hydrogens not added.
*WARNING*: Res "HID" not in HETATM Connection Database. Hydrogens not added.
*WARNING*: Res "HID" not in HETATM Connection Database. Hydrogens not added.
*WARNING*: Res "HID" not in HETATM Connection Database. Hydrogens not added.
*WARNING*: Res "HID" not in HETATM Connection Database. Hydrogens not added.
SKIPPED H( A 325 ARGHH21 ): A 325 ARG NH2 bonds- A 329 MET SD (H bumps)
*WARNING*: Res "HID" not in HETATM Connection Database. Hydrogens not added.
*WARNING*: Res "HID" not in HETATM Connection Database. Hydrogens not added.
*WARNING*: Res "HID" not in HETATM Connection Database. Hydrogens not added.
In any case, I have uploaded this other publicly available protein/DNA match here. Thank you very much
github_pdb.zip
Marc
from lightdock.
alchemistcai
commented on May 28, 2024
Preparing PDB files with pdb4amber.
A much easier way to work with amberFF's PDB files.Powered by Ambertools.
pdb4amber --reduce github_protein.pdb >gpambertools.pdb will help.
Ambertools must use --reduce flag to add hydrogens first to change correct atom names.
Ambertool's output has very different protonation from gromacs.Like this:
I'm not sure which one works better on protonation,Gromacs or Reduce.
from lightdock.
 JorgeRoel
commented on May 28, 2024
JorgeRoel
commented on May 28, 2024
Thanks @alchemistcai. Let's be patient 👼
from lightdock.
mhyleung
commented on May 28, 2024
Thank you very much!
I tried Ambertools as mentioned above, the --reduce option gave me an REDUCE returned non-zero exit status: See reduce_info.log for more details error. Let me try gromacs also and see what I get.
from lightdock.
mhyleung
commented on May 28, 2024
Dear both
I got pdb2pqr set up, but just to test I used the file kindly prepared by @alchemistcai above. I just want to make sure that I do not need to run reduce again for that "fixed" pdb file, and instead use that file directly for the lightdock3_setup.py command right? Thanks
Marc
from lightdock.
brianjimenez
commented on May 28, 2024
Hi @mhyleung , those files prepared by @alchemistcai should work. Please also note than the dna.pdb file is using HETATM instead of ATOM in your provided files. Please rename those entries before running reduce_to_amber.py or lightdock3_setup.py. Moreover, if the protein or the dna partners have missing backbone atoms, please rebuild them or just remove those nucleotides or residues before running LightDock if you enable ANM. Otherwise normal modes calculation will fail.
from lightdock.
brianjimenez
commented on May 28, 2024
After several iterations, I note down here a couple of important points:
reduce_to_amber.pyonly processesATOMentries and notHETATMin PDB files. SinceATOMis reserved for standard residues or nucleotides, this should be the expected behavior.- It is important to deal with missing atoms in backbone structure, specially when enabling flexibility through ANM.
reducecommand and many other software for managing protonation may add capping hydrogens and other atoms, which could be not supported by default in the specific scoring function used in LightDock at the time. Simply remove them if you encounter issues at the setup or simulations steps (if in the simulation, remove them from the PDB files, clean all generated files by LightDock during setup and run again setup script).- Empty chains are not supported by LightDock, indeed, empty chains might be a bad practice when dealing with PDB information.
I close down for the moment this issue, please feel free to reopen it if there are more questions or issues related to the topic.
from lightdock.
Related Issues (20)
- what the datas meaning of cluster.repr file? HOT 4
- Unexpected keyword argument 'seed' HOT 6
- lightdock pdb file visualises incorrectly in pymol HOT 5
- ERROR: [NormalModesCalculationError] Number of atoms is different HOT 6
- Swarm number remains 400 after using residue restraints HOT 5
- Can't find implicit membrane model for PBP2a protein on Memdock. HOT 6
- Issues when docking HOT 1
- test_dna and test_pydock failed. HOT 2
- TOBI scoring function fails to replicate results with CCharPPI server HOT 5
- Error encountered with DNA scoring function in lightdock3.py for DNA-DNA docking HOT 3
- Migrate tests from nose to pytest HOT 2
- Question about the treatment of protein-ligand docking. HOT 3
- Freezes after a while HOT 6
- Support for non standard amino acids HOT 2
- Ligands are not placed in designated swarm centers HOT 6
- Performance for protein-DNA docking HOT 1
- Use MPI for simulation HOT 3
- Much larger number of swarms in restraints tutorial than shown in docs HOT 4
- [lgd_cluster_bsas.py] Clustering has failed. HOT 9
Recommend Projects
-
 React
React
A declarative, efficient, and flexible JavaScript library for building user interfaces.
-
Vue.js
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
-
 Typescript
Typescript
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
-
TensorFlow
An Open Source Machine Learning Framework for Everyone
-
Django
The Web framework for perfectionists with deadlines.
-
 Laravel
Laravel
A PHP framework for web artisans
-
D3
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
-
Recommend Topics
-
javascript
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
-
web
Some thing interesting about web. New door for the world.
-
server
A server is a program made to process requests and deliver data to clients.
-
Machine learning
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
-
Visualization
Some thing interesting about visualization, use data art
-
Game
Some thing interesting about game, make everyone happy.
Recommend Org
-
Facebook
We are working to build community through open source technology. NB: members must have two-factor auth.
-
Microsoft
Open source projects and samples from Microsoft.
-
Google
Google ❤️ Open Source for everyone.
-
Alibaba
Alibaba Open Source for everyone
-
D3
Data-Driven Documents codes.
-
Tencent
China tencent open source team.
from lightdock.