This package is a integrated Defect Formation energy package, which contains generating tetrahedral interstitial sites and octahedral interstitial sites, submitting VASP calculation job and extracting necessary data to calculate defect formation energy.
- Defect-Formation-Calculation
Firstly, you should make a directory for those scripts and add its path to your .bashrc. I divide these scripts into three directories so that you can easily know the usage of each scripts, and you can put them all into a directory in order to conveniently use them. You can also maintain the configuration without any change, so you have to add all file path to $PATH so that you can execute them.
For example, I make the scripts directory under my $HOME directory, so I can use the below command to add this directory to $PATH, and source your .bashrc to make it work.
echo export PATH="$PATH:/home/${usr_name}/scripts" >> ~/.bashrc
source ~/.bashrcNoted that, if you get permission denied, please use chmod u+x pyvasp.py to give the execute right to the file
There are three kinds of defect system you can generate, vacancy defect, purity defect and interstitial defect. All these can be generated by defectmaker.py, and the generated structures in VASP format will be stored in a directory named by its attributes.
Here we supply the command pyvasp.py to generate the defect POSCAR you want.
module load sagar #load the necesary package
pyvasp.py --help # you can get some short help from this command
pyvasp.py main --help # get the help of a specific command Follow the below example, the parameter -i means the the atom which will dopped into the system, the parameter -o means the atom which will be removed.
pyvasp.py get_purity --help # get some help
# generate purity defect poscar including vacancy defect
# this means generate a Si-vacancy defect
pyvasp.py get_purity_poscar supcell.vasp -i Vacc -o Si
# generate H tetrahedral sites defect
pyvasp.py get_tetrahedral POSCAR -i HFirst, we supply some shell scripts to generate those input file for VASP calculation, they are potcar.sh, incar.sh, kpoints.sh. Below I will simply introduce the usage of these scripts.
These scripts will not work if correspondent files exist, for example, incar.sh will not work if an INCAR file exists in your current directory
There are two parameters you can input. The first parameter is the type of potcar
- 1 is correspondent to
PAW_PBE - 2 is correspondent to
PAW_LDA - 3 is correspondent to
PAW_PW91 - 4 is correspondent to
USPP_LDA - 5 is correspondent to
USPP_PW91
The defaul of first parameter is 1, and if it can uncompress POTCAR.Z file to POTCAR.
The second parameter is the type of atom potcar, maybe the same atom has some different type of potcar, for example, Mg atom has Mg, Mg_GW, Mg_pv, Mg_pv_GW, Mg_sv, Mg_sv_GW potcar, so you can specify one of them to get specific POTCAR, the default of this parameter is the atom itself.
potcar.sh 2 # this will generate the POTCAR based on your PSOCAR from PAW_LDA directory
potcar.sh 1 Mg_pv # noted that 1 can be omitedThis script is used to generate INCAR for your system, and it supposes that the POTCAR has been in your directory, because ENCUT should be set based on the ENMAX of your POTCAR.
This script is used to generate KPOINTS, the usage is:
kpoint.sh 40 # this will generate KPOINTS mesh 40/a 40/b 40/c
kpoint.sh band # this will genrate k-path based on `aflow`- kp_test.sh can test what kinds of KPOINTS you should use
- encut_test.sh can help you test what
ENCUTis best suitable for your calculations. - latt_const_test.sh can test which lattice constant is best for the system you are calculating.
Here, we supply some integrated shell scripts to calculate the jobs you need.
- stru_relax.sh (structure relax, ISIF=3)
- stru_optimization.sh (structure optimization, ISIF=2)
- stru_scf.sh (structure self consistent field calculation)
- stru_band.sh (band calculation)
- stru_dos.sh (density of states calculation)
- job.sh (submit your job)
Here, almost all *.sh file will automatic make a new directory for its calculation, and will arrange in the top directory in parallel except for stru_relax.sh and stru_optimization.sh, because we need CONTCAR from relax or optimization when doing self-consisten-field calculation, if we make a single directory for relax and optimization, then you should specify the path of CONTCAR(whether under relax or optimization), so here we do not make a single directory for relax and optimization.
And each file can be executed respectively with some necessary files. Below I list the necessary files each script need to read in the current directory.
stru_relax,stru_optimizationneed POSCARstru_scfneeds CONTCAR, POTCAR, INCARstru_dos,stru_bandneeds scf/CHG* scf/WAVECAR scf/INCAR scf/POTCAR scf/KPOINTS scf/CONTCARstru_bandneeds scf/WAVECAR scf/CHG* scf/POTCAR scf/POSCAR scf/INCAR
Below is an example.
[hecc@cmp Si]$ ll
total 416
drwxrwxr-x 2 hecc hecc 272 Apr 4 15:20 band
-rw-rw-r-- 1 hecc hecc 0 Apr 4 15:19 CHG
-rw-rw-r-- 1 hecc hecc 0 Apr 4 15:19 CHGCAR
-rw-rw-r-- 1 hecc hecc 512 Apr 4 15:19 CONTCAR
drwxrwxr-x 2 hecc hecc 4096 Apr 4 15:20 dos
-rw-rw-r-- 1 hecc hecc 11073 Apr 4 15:19 DOSCAR
-rw-rw-r-- 1 hecc hecc 15562 Apr 4 15:19 EIGENVAL
-rw-rw-r-- 1 hecc hecc 3582 Apr 4 15:19 IBZKPT
-rw-rw-r-- 1 hecc hecc 152 Apr 4 15:19 INCAR
-rwxrw-r-- 1 hecc hecc 234 Apr 4 15:19 job.sh
-rw-rw-r-- 1 hecc hecc 27 Apr 4 15:19 KPOINTS
-rw-rw-r-- 1 hecc hecc 1180 Apr 4 15:19 OSZICAR
-rw-rw-r-- 1 hecc hecc 77692 Apr 4 15:19 OUTCAR
-rw-rw-r-- 1 hecc hecc 234 Apr 4 15:19 PCDAT
-rw-rw-r-- 1 hecc hecc 345 Apr 4 15:19 POSCAR
-rw-rw-r-- 1 hecc hecc 195673 Apr 4 15:19 POTCAR
-rw-rw-r-- 1 hecc hecc 0 Apr 4 15:19 REPORT
drwxrwxr-x 2 hecc hecc 323 Apr 4 15:19 scf
-rw-rw-r-- 1 hecc hecc 17026 Apr 4 15:20 slurm-54254.out
-rw-rw-r-- 1 hecc hecc 57847 Apr 4 15:19 vasprun.xml
-rw-rw-r-- 1 hecc hecc 0 Apr 4 15:19 WAVECAR
-rw-rw-r-- 1 hecc hecc 283 Apr 4 15:19 XDATCAR
We also supply some scripts to generate the input files needed in VASP Calculation, such as INCAR, KPOINTS, POTCAR, you can also use these scripts to generate them. So in general, you can just begin your job from a POSCAR and a job.sh.
Here we supply a command interface to get the value you want.
Noted that, if you get permission denied, please use chmod u+x pyvasp.py to give the execute right to the file
module load sagar #load the necesary package
pyvasp.py --help # you can get some short help from this command
pyvasp.py main --help # get the help of a specific command This command is used to get some common value of your calculation system. For instance, gap, fermi energy, electrons number and so on. The last parameter is the directory path of your calculation system, make sure it is right or you will get wrong answer.
pyvasp.py main -a gap . # this can read the gap and vbm, cbm
pyvasp.py main -a fermi . # this can read the fermi energy
pyvasp.py main -a energy . # this can read the total energy
pyvasp.py main -a ele . # this can read the electrons in your OUTCAR
pyvasp.py main -a ele-free . # this can get electrons number of the defect-free system
pyvasp.py main -a image image_corr/ # this can get Ewald energy of your system, using `pyvasp.py main -a ewald image_corr` can also get the same result.This command is used to extend your cell and generate a supcell.vasp
pyvasp.py cell -v 2 2 2 POSCAR
# extend your POSCAR to 2*2*2 supercellThis command is used to get the purity structures , such Si-vacancy, Ga purity in In2O3 system, but noted that each time only one purity atom will be dopped into the system.
pyvasp.py get_purity -i Vacc -o Si Si-POSCAR # generate a vacancy
pyvasp.py get_purity -i Ga -o In In2O3-POSCAR #genrate a Ga defectThis command is used to get the tetrahedral interstitial sites, for example, in YFe2 system, H atom can be inserted into the tetrahedral sites.
pyvasp.py get_tetrahedral -i H YFe2-POSCARThis command can get the electrostatic of your defect system and no defect system of the farther atom from defect atom
pyvasp.py get_PA defect_free charge_state_1This command can get some symmetry message of your POSCAR.
pyvasp.py symmetry -a spacegroup POSCAR # get space group
pyvasp.py symmetry -a equivalent POSCAR # get equivalent atoms
pyvasp.py symmetry -a primitive POSCAR # generate primitive cell POSCARThis command can generate the chemical potential phase figure, noted that we only support three-component compound so that we can plot a two dimension figure.
pyvasp.py chem_pot chemical-incar
pyvasp.py chem_pot chemical-incar -r 2
# remove the second dimensionHere I will give some examples to demonstrate how this package works
Prepare your POSCAR in your work directory, if you want to use the default setting, you can just execute the command like stru_relax.sh to calculate your system for which can generate INCAR, KPOINTS, POTCAR automatic.
#!/bin/bash -l
#NOTE the -l flag!
#SBATCH -J job-name
#SBATCH -p super_q -N 1 -n 12
#SBATCH -t 10-0:0:0
# NOTE Each small node has 12 cores
#
export NSLOTS=$SLURM_NPROCS
module load vasp/5.4.4-impi-mkl
stru_relax.sh
stru_scf.sh 45 # using k-mesh 45/a 45/b 45/c
stru_band.sh 20 # insert 20 pts between two high-symmetry pts
stru_dos.sh 50 #using k-mesh 50/a 50/b 50/cThe initial files can only be POSCAR and job.sh
[hecc@cmp Si]$ ll
total 8
-rwxrw-r-- 1 hecc hecc 234 Apr 4 15:19 job.sh
-rw-rw-r-- 1 hecc hecc 345 Apr 4 15:19 POSCAR
then you can sbath your job, and after calculation you will get the below files.
[hecc@cmp Si]$ ll
total 416
drwxrwxr-x 2 hecc hecc 272 Apr 4 15:49 band
-rw-rw-r-- 1 hecc hecc 0 Apr 4 15:49 CHG
-rw-rw-r-- 1 hecc hecc 0 Apr 4 15:49 CHGCAR
-rw-rw-r-- 1 hecc hecc 512 Apr 4 15:49 CONTCAR
drwxrwxr-x 2 hecc hecc 4096 Apr 4 15:49 dos
-rw-rw-r-- 1 hecc hecc 11073 Apr 4 15:49 DOSCAR
-rw-rw-r-- 1 hecc hecc 15562 Apr 4 15:49 EIGENVAL
-rw-rw-r-- 1 hecc hecc 3582 Apr 4 15:49 IBZKPT
-rw-rw-r-- 1 hecc hecc 152 Apr 4 15:49 INCAR
-rwxrw-r-- 1 hecc hecc 234 Apr 4 15:19 job.sh
-rw-rw-r-- 1 hecc hecc 27 Apr 4 15:49 KPOINTS
-rw-rw-r-- 1 hecc hecc 1180 Apr 4 15:49 OSZICAR
-rw-rw-r-- 1 hecc hecc 77692 Apr 4 15:49 OUTCAR
-rw-rw-r-- 1 hecc hecc 234 Apr 4 15:49 PCDAT
-rw-rw-r-- 1 hecc hecc 345 Apr 4 15:49 POSCAR
-rw-rw-r-- 1 hecc hecc 195673 Apr 4 15:49 POTCAR
-rw-rw-r-- 1 hecc hecc 0 Apr 4 15:49 REPORT
drwxrwxr-x 2 hecc hecc 323 Apr 4 15:49 scf
-rw-rw-r-- 1 hecc hecc 57847 Apr 4 15:49 vasprun.xml
-rw-rw-r-- 1 hecc hecc 0 Apr 4 15:49 WAVECAR
-rw-rw-r-- 1 hecc hecc 283 Apr 4 15:49 XDATCAR
Here, the main directory contains relax-results or optimization-results, the band, dos, scf will respectively contain correspondent files.
First, you should supply the POSCAR of Si, and execute
# generate Si-vacancy structures
pyvasp.py get_purity -i Vacc -o Si POSCARto generate purity POSCAR, and then submit your job. Below is an example job file.
#!/bin/bash -l
# NOTE the -l flag!
#
#SBATCH -J Si
# Default in slurm
# Request 5 hours run time
#SBATCH -t 4-5:0:0
#
#SBATCH -p super_q -N 1 -n 12
# NOTE Each small node has 12 cores
#
module load vasp/5.4.4-impi-mkl
# add your job logical here!!!
# this is the defect directory
defect_folder=Si-Vacc-defect
export NSLOTS=$SLURM_NPROCS
mkdir supercell
cp POSCAR supercell/
cd supercell
stru_relax.sh
stru_scf.sh
cd ..
get_ground_defect_stru.sh $defect_folder
cd $defect_folder
for q in -2 -1 0 1 2
do
charge_state_cal.sh $q
done
cd ..
image_corr_cal.shYou can get a standard hierarchy of files if you do not encounter any accident problems after all calculations have been completed. Below is an example, and your files should also be like this.
[hecc@cmp ~]$ tree Si/ -d
Si/
├── image_corr
├── Si-Vacc-defect
│ ├── charge_state_0
│ │ └── scf
│ ├── charge_state_1
│ │ └── scf
│ ├── charge_state_-1
│ │ └── scf
│ ├── charge_state_2
│ │ └── scf
│ ├── charge_state_-2
│ │ └── scf
│ └── POSCAR-Si-Vacc-defect_id0.vasp-dir
└── supercell
└── scf
Next, you can execute defect_formation_energy.py
to plot the figure. But before you push your enter key, you should supply a defect-incar including epsilon(dielectric coefficient) and the chemical potential of some elements in your current directory. The defect-incar may be like this
epsilon=13.36
mu_Si = -5.41
then you can push your enter key and wait for the defect-formation energy figure.
# the first parameter is the path of main directory
# the second parameter is the path of your defect directory
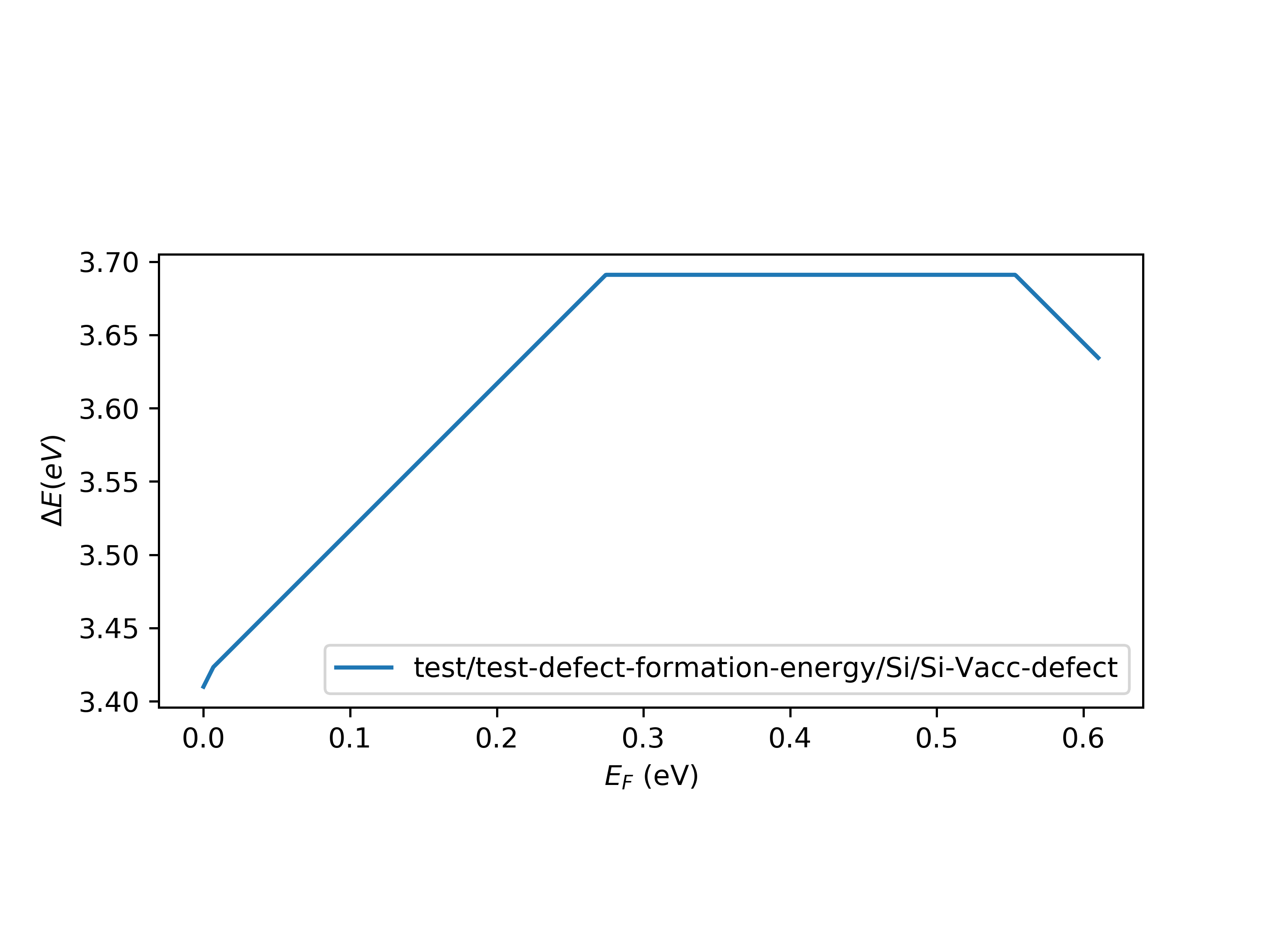
defect_formation_energy.py Si Si/Si-Vacc-defectAlso, we will write a log file named ${defect_folder}_log.txt to record some necessary message in your calculation process. Below is an example of log-file of Si-vacancy defect, you can obtain some useful information from this log-file.
test/test-defect-formation-energy/Si/Si-Vacc-defect
Energy of supcell is: -1171.6512 eV
Evbm: 5.4746749999999995 eV
Ecbm: 6.084886 eV
gap: 0.6102110000000005 eV
charge energy E_PA E_IC far_atom_def_sys far_atom_def_fr_system
0 -1162.55020 -0.00000 +0.00000 35 35
-2 -1150.69310 +0.21600 +0.24860 35 35
2 -1173.95500 -0.07420 +0.24860 35 35
-1 -1156.67410 +0.09000 +0.06215 35 35
1 -1168.31110 -0.05030 +0.06215 35 35
chemical potential of Si: -5.41 eV
chemical potential of Vacc: 0 eV
Si has been removed
Vacc has been doped
This is the defect formation energy figure.
For multiple composition, you should get the proper chemical potential of some elements. You should supply a chemical-incar. The left is the the composition and the right is the energy. Noted that the host compound should be marked with #, and you should supply the pure phase energy.
Ga=-2.916203375
Ga8O12=-121.098
O2=-8.9573588
Zn=-2.5493
#Zn8Ga16O32=-328.32564
ZnO=-10.586057
And use the command
python3.6 pyvasp.py chem_pot -r 0 chemical-incar
you can get the cross points in chemical_log.txt file.
chemical potential constrain of Ga8O12
Zn Ga O
-3.7382 -5.503 0.0
-0.0695 0.0 -3.6687
0.0 0.1043 -3.7382
chemical potential constrain of ZnO
Zn Ga O
-3.5581 -5.5931 0.0
0.1707 0.0 -3.7287
0.0 -0.256 -3.5581
and also a chemical potential phase figure will be generated.




