This repository has employed the following pipelines in order to arrive at a consensus on drug-resistant Mycobacterium tuberculosis predictions:
-
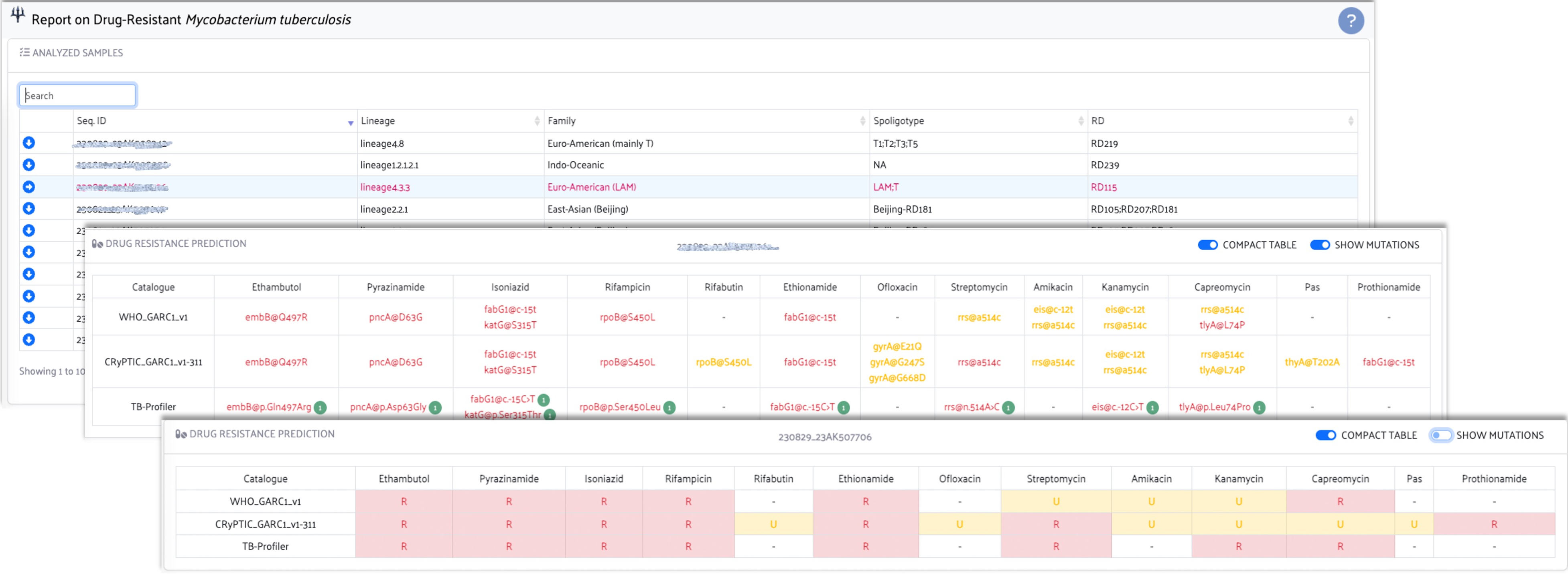
CRyPTIC: The pipeline involves decontaminating reads, performing quality controls and variant calling using various tools and the the H37Rv reference genome. We have incorporated the pipeline into the nextflow DSL2 and employed precompiled reference genome databases (e.g., H37Rv, nontuberculous Mycobacterium, human, etc.) to facilitate analyzing input genomes. Moreover, the gnomonicus repository is used to predict durg reistance based on the detected mutations and catalogues available. The spear-MTB utilizes WHO and CRyPTIC catalogues, which are presented in GARC1 format and maintained here.
-
TB Profiler: The pipeline includes aligning reads to the H37Rv reference genome, utilizing a pairwise aligner and then identifying variations with the use of bcftools. The catalogue of mutations is in hgvs nomenclature and is maintained here.
⚠ Warning: The spear-MTB pipeline is tested using Illumina paired-end reads and on Unix-based system having Singularity.
Befor running the pipeline, please ensure that you have the following programs installed:
- Nextflow
- Singularity
- conda
After the successful installations, proceed with running the following commands:
git clone https://github.com/xrazmo/spear-mtb.git
cd ./spear-mtb | chmod +x ./setup.sh | ./setup.sh
These commands perform the following steps:
- Downloading the repository from Github.
- Creating a conda environment named spear-mtb.
- Downloading and extracting the precompiled assets, which encompasses indexed genomes, the reference H37rv, mutations catalogues, and a KRAKEN2 database containing genomes belong to Mycobacteriaceae family.
For updating mutation catalogues run the following command:
./setup.sh -u
Before running the pipeline, please review and modify the configuration file (nextflow.config) to suit your specific requirements and preferences. Note that the current configuration file includes predefined profiles and resource allocation designed for the RACKHAM cluster at UPPMAX.
Usage:
spear-mtb.sh [-t <tmp_dir>] [-c <config_file>] [-a <assets_dir>] [-p <prefix>] [-f <profile>] [-o <out_dir>] <input_directory>
Options:
-t Temporary directory: Set the location for temporary files (default: PARENT_DIR/.tmp)
-c Config file: Specify a Nextflow configuration file (default: nextflow.config)
-a Assets directory: Specify the assets directory containing reference genomes and catalogues (default: SCRIPT_DIR/assets)
-p Prefix: Customize the name of the trace file produced by Nextflow (default: date-time).
-f Profile: Specify the profile already defined in your config file (default: 'slurm')
-o Output directory: output directory containing results: vcf, csv, json, html files (default: PARENT_DIR/out)
Arguments:
<input_directory> Input directory: specify a directory containing Illumina paired-end reads or folders containing them.
Examples:
spear-mtb.sh path/to/input_data
spear-mtb.sh -t /tmp -c custom.config -a /path/to/assets -p myprefix -f slurm -o /path/to/output /path/to/input_data
SPEAR-MTB integrates predictions based on different pipelines and catalogues in an interactive offline HTML file, which will be delivered to your specified output directory. For more information, please refer to the help window within the report.