![]()



![]()

Website | Nature Chemical Biology 2022 Paper | NeurIPS 2021 Paper | Long Paper | Slack | TDC Mailing List | TDC Documentation | Contribution Guidelines
Artificial intelligence is poised to reshape therapeutic science. Therapeutics Data Commons is a coordinated initiative to access and evaluate artificial intelligence capability across therapeutic modalities and stages of discovery, supporting the development of AI methods, with a strong bent towards establishing the foundation of which AI methods are most suitable for drug discovery applications and why.
Researchers across disciplines can use TDC for numerous applications. AI-solvable tasks, AI-ready datasets, and curated benchmarks in TDC serve as a meeting point between biochemical and AI scientists. TDC facilitates algorithmic and scientific advances and accelerates machine learning method development, validation, and transition into biomedical and clinical implementation.
TDC is an open-science initiative. We welcome contributions from the community.
[1] Velez-Arce, Huang, Li, Lin, et al., TDC-2: Multimodal Foundation for Therapeutic Science, bioRxiv, 2024 [Paper]
[2] Huang, Fu, Gao, et al., Artificial Intelligence Foundation for Therapeutic Science, Nature Chemical Biology, 2022 [Paper]
[3] Huang, Fu, Gao, et al., Therapeutics Data Commons: Machine Learning Datasets and Tasks for Drug Discovery and Development, NeurIPS 2021 [Paper] [Poster]
[4] Huang et al., Benchmarking Molecular Machine Learning in Therapeutics Data Commons, ELLIS ML4Molecules 2021 [Paper] [Slides]
[5] Huang et al., Therapeutics Data Commons: Machine Learning Datasets and Tasks for Drug Discovery and Development, Baylearn 2021 [Slides] [Poster]
[6] Huang, Fu, Gao et al., Therapeutics Data Commons, NSF-Harvard Symposium on Drugs for Future Pandemics 2020 [#futuretx20] [Slides] [Video]
[7] TDC User Group Meetup, Jan 2022 [Agenda]
[8] Zitnik, Machine Learning to Translate the Cancer Genome and Epigenome Session, AACR Annual Meeting 2022, Apr 2022
[9] Zitnik, Few-Shot Learning for Network Biology, Keynote at KDD Workshop on Data Mining in Bioinformatics
[10] Zitnik, Actionable machine learning for drug discovery and development, Broad Institute, Models, Inference & Algorithms Seminar, 2021
[11] Zitnik, Graph Neural Networks for Biomedical Data, Machine Learning in Computational Biology, 2020
[12] Zitnik, Graph Neural Networks for Identifying COVID-19 Drug Repurposing Opportunities, MIT AI Cures, 2020
- Diverse areas of therapeutics development: TDC covers a wide range of learning tasks, including target discovery, activity screening, efficacy, safety, and manufacturing across biomedical products, including small molecules, antibodies, and vaccines.
- Ready-to-use datasets: TDC is minimally dependent on external packages. Any TDC dataset can be retrieved using only 3 lines of code.
- Data functions: TDC provides extensive data functions, including data evaluators, meaningful data splits, data processors, and molecule generation oracles.
- Leaderboards: TDC provides benchmarks for fair model comparison and systematic model development and evaluation.
- Open-source initiative: TDC is an open-source initiative. If you want to get involved, let us know.
See here for the latest updates in TDC!
To install the core environment dependencies of TDC, use pip:
pip install PyTDCNote: TDC is in the beta release. Please update your local copy regularly by
pip install PyTDC --upgradeThe core data loaders are lightweight with minimum dependency on external packages:
numpy, pandas, tqdm, scikit-learn, fuzzywuzzy, seabornFor utilities requiring extra dependencies, TDC prints installation instructions. To install full dependencies, please use the following conda-forge solution.
Data functions for molecule oracles, scaffold split, etc., require certain packages like RDKit. To install those packages, use the following conda installation:
conda install -c conda-forge pytdcWe provide tutorials to get started with TDC:
| Name | Description |
|---|---|
| 101 | Introduce TDC Data Loaders |
| 102 | Introduce TDC Data Functions |
| 103.1 | Walk through TDC Small Molecule Datasets |
| 103.2 | Walk through TDC Biologics Datasets |
| 104 | Generate 21 ADME ML Predictors with 15 Lines of Code |
| 105 | Molecule Generation Oracles |
| 106 | Benchmark submission |
| DGL | Demo presented at DGL GNN User Group Meeting |
| U1.1 | Demo presented at first TDC User Group Meetup |
| U1.2 | Demo presented at first TDC User Group Meetup |
| 201 | TDC-2 Resource and Multi-modal Single-Cell API |
| 202 | TDC-2 Resource and PrimeKG |
| 203 | TDC-2 Resource and External APIs |
| 204 | TDC-2 Model Hub |
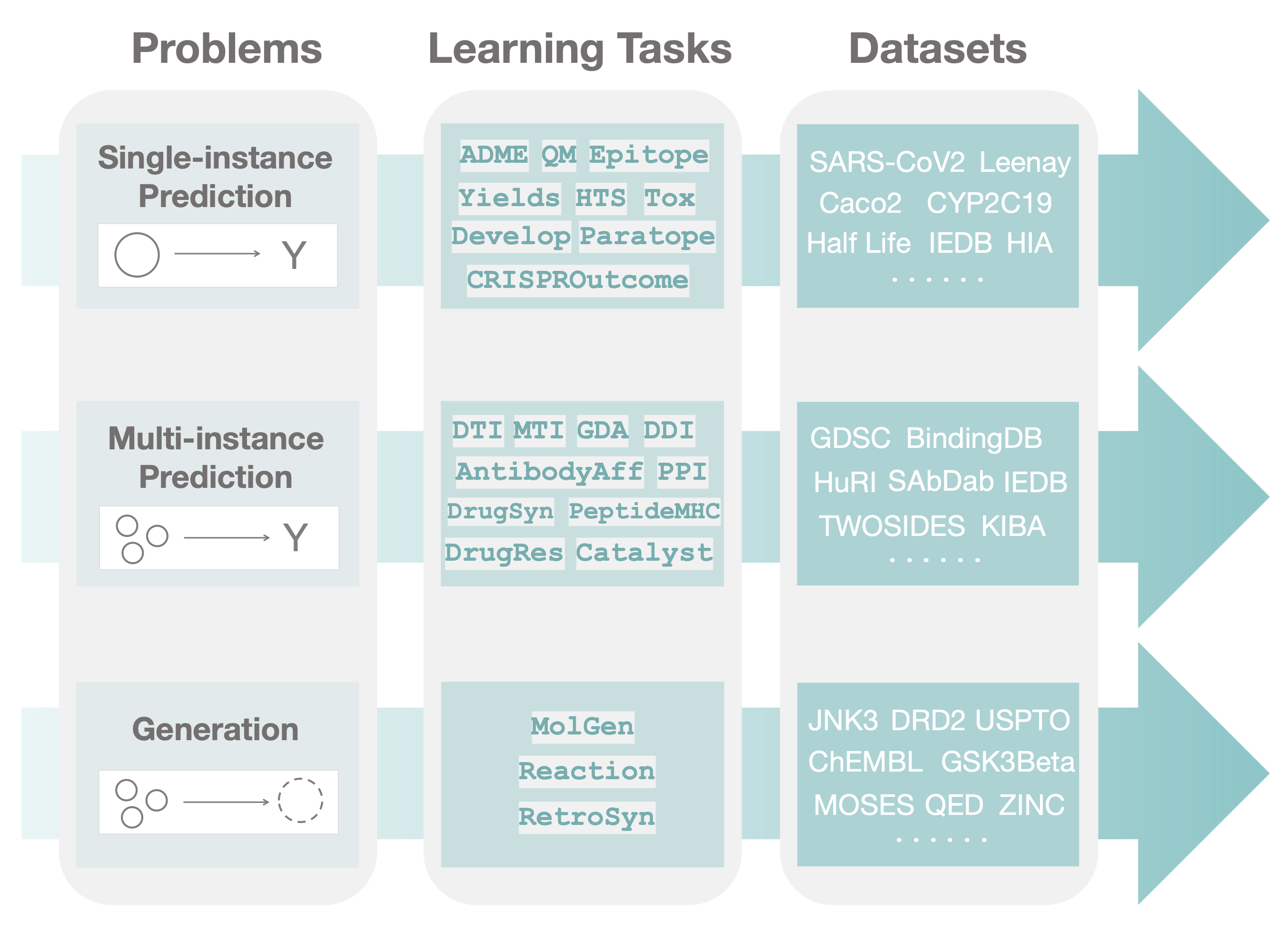
TDC has a unique three-tiered hierarchical structure, which to our knowledge, is the first attempt at systematically organizing machine learning for therapeutics. We organize TDC into three distinct problems. For each problem, we give a collection of learning tasks. Finally, for each task, we provide a series of datasets.
In the first tier, after observing a large set of therapeutics tasks, we categorize and abstract out three major areas (i.e., problems) where machine learning can facilitate scientific advances, namely, single-instance prediction, multi-instance prediction, and generation:
- Single-instance prediction
single_pred: Prediction of property given individual biomedical entity. - Multi-instance prediction
multi_pred: Prediction of property given multiple biomedical entities. - Generation
generation: Generation of new desirable biomedical entities.

The second tier in the TDC structure is organized into learning tasks. Improvement in these tasks can result in numerous applications, including identifying personalized combinatorial therapies, designing novel classes of antibodies, improving disease diagnosis, and finding new cures for emerging diseases.
Finally, in the third tier of TDC, each task is instantiated via multiple datasets. For each dataset, we provide several splits of the dataset into training, validation, and test sets to simulate the type of understanding and generalization (e.g., the model's ability to generalize to entirely unseen compounds or to granularly resolve patient response to a polytherapy) needed for transition into production and clinical implementation.
TDC provides a collection of workflows with intuitive, high-level APIs for both beginners and experts to create machine learning models in Python. Building off the modularized "Problem -- Learning Task -- Data Set" structure (see above) in TDC, we provide a three-layer API to access any learning task and dataset. This hierarchical API design allows us to easily incorporate new tasks and datasets.
For a concrete example, to obtain the HIA dataset from the ADME therapeutic learning task in the single-instance prediction problem:
from tdc.single_pred import ADME
data = ADME(name = 'HIA_Hou')
# split into train/val/test with scaffold split methods
split = data.get_split(method = 'scaffold')
# get the entire data in the various formats
data.get_data(format = 'df')You can see all the datasets that belong to a task as follows:
from tdc.utils import retrieve_dataset_names
retrieve_dataset_names('ADME')See all therapeutic tasks and datasets on the TDC website!
To retrieve the training/validation/test dataset split, you could simply type
data = X(name = Y)
data.get_split(seed = 42)
# {'train': df_train, 'val': df_val, 'test': df_test}You can specify the splitting method, random seed, and split fractions in the function by e.g. data.get_split(method = 'scaffold', seed = 1, frac = [0.7, 0.1, 0.2]). Check out the data split page on the website for details.
We provide various evaluation metrics for the tasks in TDC, which are described in model evaluation page on the website. For example, to use metric ROC-AUC, you could simply type
from tdc import Evaluator
evaluator = Evaluator(name = 'ROC-AUC')
score = evaluator(y_true, y_pred)TDC provides numerous data processing functions, including label transformation, data balancing, pairing data to PyG/DGL graphs, negative sampling, database querying, and so on. For function usage, see our data processing page on the TDC website.
For molecule generation tasks, we provide 10+ oracles for both goal-oriented and distribution learning. For detailed usage of each oracle, please check out the oracle page on the website. For example, we want to retrieve the GSK3Beta oracle:
from tdc import Oracle
oracle = Oracle(name = 'GSK3B')
oracle(['CC(C)(C)....'
'C[C@@H]1....',
'CCNC(=O)....',
'C[C@@H]1....'])
# [0.03, 0.02, 0.0, 0.1]Every dataset in TDC is a benchmark, and we provide training/validation and test sets for it, together with data splits and performance evaluation metrics. To participate in the leaderboard for a specific benchmark, follow these steps:
-
Use the TDC benchmark data loader to retrieve the benchmark.
-
Use training and/or validation set to train your model.
-
Use the TDC model evaluator to calculate the performance of your model on the test set.
-
Submit the test set performance to a TDC leaderboard.
As many datasets share a therapeutics theme, we organize benchmarks into meaningfully defined groups, which we refer to as benchmark groups. Datasets and tasks within a benchmark group are carefully curated and centered around a theme (for example, TDC contains a benchmark group to support ML predictions of the ADMET properties). While every benchmark group consists of multiple benchmarks, it is possible to separately submit results for each benchmark in the group. Here is the code framework to access the benchmarks:
from tdc import BenchmarkGroup
group = BenchmarkGroup(name = 'ADMET_Group', path = 'data/')
predictions_list = []
for seed in [1, 2, 3, 4, 5]:
benchmark = group.get('Caco2_Wang')
# all benchmark names in a benchmark group are stored in group.dataset_names
predictions = {}
name = benchmark['name']
train_val, test = benchmark['train_val'], benchmark['test']
train, valid = group.get_train_valid_split(benchmark = name, split_type = 'default', seed = seed)
# --------------------------------------------- #
# Train your model using train, valid, test #
# Save test prediction in y_pred_test variable #
# --------------------------------------------- #
predictions[name] = y_pred_test
predictions_list.append(predictions)
results = group.evaluate_many(predictions_list)
# {'caco2_wang': [6.328, 0.101]}For more information, visit here.
If you find Therapeutics Data Commons useful, cite our latest preprint, our NeurIPS paper, and Nature Chemical Biology paper :
@article {Velez-Arce2024tdc,
author = {Velez-Arce, Alejandro and Huang, Kexin and Li, Michelle and Lin, Xiang and Gao, Wenhao and Fu, Tianfan and Kellis, Manolis and Pentelute, Bradley L. and Zitnik, Marinka},
title = {TDC-2: Multimodal Foundation for Therapeutic Science},
elocation-id = {2024.06.12.598655},
year = {2024},
doi = {10.1101/2024.06.12.598655},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/early/2024/06/21/2024.06.12.598655},
eprint = {https://www.biorxiv.org/content/early/2024/06/21/2024.06.12.598655.full.pdf},
journal = {bioRxiv}
}
@article{Huang2021tdc,
title={Therapeutics Data Commons: Machine Learning Datasets and Tasks for Drug Discovery and Development},
author={Huang, Kexin and Fu, Tianfan and Gao, Wenhao and Zhao, Yue and Roohani, Yusuf and Leskovec, Jure and Coley,
Connor W and Xiao, Cao and Sun, Jimeng and Zitnik, Marinka},
journal={Proceedings of Neural Information Processing Systems, NeurIPS Datasets and Benchmarks},
year={2021}
}
@article{Huang2022artificial,
title={Artificial intelligence foundation for therapeutic science},
author={Huang, Kexin and Fu, Tianfan and Gao, Wenhao and Zhao, Yue and Roohani, Yusuf and Leskovec, Jure and Coley,
Connor W and Xiao, Cao and Sun, Jimeng and Zitnik, Marinka},
journal={Nature Chemical Biology},
year={2022}
}
TDC is built on top of other open-sourced projects. If you used these datasets/functions in your research, please cite the original work as well. You can find the original paper on the website for the function/dataset.
TDC is a community-driven and open-science initiative. To get involved, join our Slack Workspace and check out the contribution guide!
Reach us at [email protected] or open a GitHub issue.
TDC is hosted on Harvard Dataverse with the following persistent identifier https://doi.org/10.7910/DVN/21LKWG. When Dataverse is under maintenance, TDC datasets cannot be retrieved. That happens rarely; please check the status on the Dataverse website.
TDC codebase is under MIT license. For individual dataset usage, please refer to the dataset license found in the website.


















































































































































