Discovery of significant mutations around splice sites in differentially expressed genes in Chordoma transcriptome data using machine learning tools.
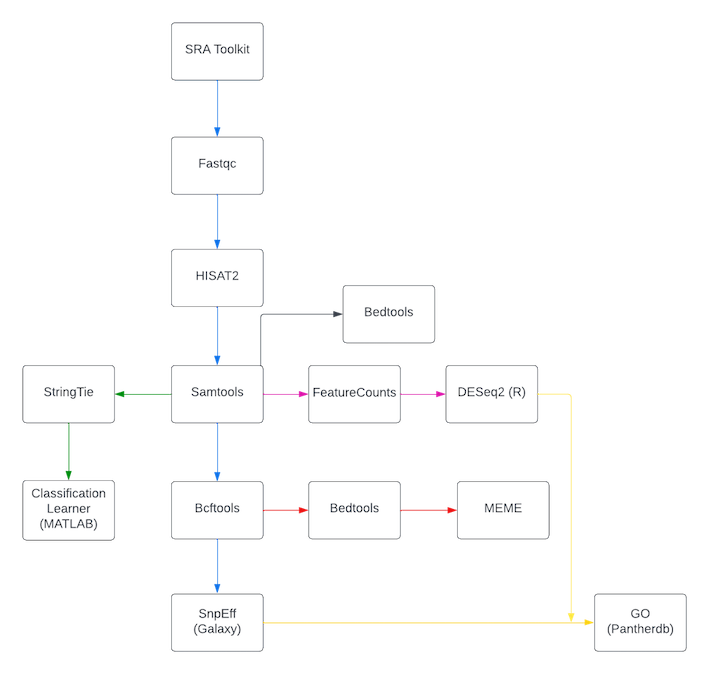
(the black line which represents intronic coverage workflow, should be started from bcftools)
Several commands that were used in this project can be seen below.
prefetch <"SAMPLE GEO CODE">
fasterq-dump --split-files <"SAMPLE GEO CODE">
hisat2 -q -x <ref.fa> -1 <fastq_1> -2 <fastq_2> -S <file.sam>
samtools view -b -o <file.bam> <file.sam>
samtools sort -o <file.sorted.bam> <file.bam>
samtools index <file.sorted.bam >
samtools mpileup -g -f <ref.fa> <file.sorted.bam> > <file.raw.bcf>
samtools view -h <sample.sorted.bam> <gene coordinates> -o <gene-name_sample-name.bam>
samtools faidx <ref.fa>
bcftools call -O b -vc <file.raw.bcf> > <file.var.bcf>
bcftools view <file.var.bcf> | vcfutils.pl varFilter - > <file.var-final.vcf>
bcftools consensus --fasta-ref <ref.fa> <file.vcf.gz> -o file.fasta
featureCounts -T 8 -a <file.gtf> "transcrip_id" -o readCounts.txt <file.bam>
stringtie -p 8 -G <file.gtf> -e -B -o <file.gtf> -A abundances.tsv <file.bam>
bedtools coverage -a <file.bed> -b <file.bam> -bed -d > file.txt
bedtools getfasta -fi <file.fasta> -bed <file.bed> -fo file.fasta
Data can be downloaded via https://drive.google.com/drive/folders/19bvqcYv53lFGFoX2HsNporkIXiE5nYmo?usp=sharing. Also, raw data is accessible here, https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP109781&o=bases_l%3Ad%3Bacc_s%3Aa. For the reference genome, http://ftp.ensembl.org/pub/release-106/fasta/homo_sapiens/dna_index/
