#!/bin/bash
#Usage: sh allhic_pbs.sh QMg_NbQ4P_RN.fasta 38 GATC
cat << EOF |qsub
#!/bin/bash -l
#PBS -N allhic
#PBS -l walltime=150:00:00
#PBS -j oe
#PBS -l mem=200G
#PBS -l ncpus=8
#PBS -l cpuarch=avx2
#PBS -M [email protected]
##PBS -m bea
cd \$PBS_O_WORKDIR
conda activate allhic
export PATH=/work/waterhouse_team/apps/ALLHiC/bin:/work/waterhouse_team/apps/ALLHiC/scripts:$PATH
echo "samtools merge and index"
#samtools merge all.hic.sorted.dedup.bam *.bam
#samtools index all.hic.sorted.dedup.bam
echo "extract"
allhic extract all.hic.sorted.dedup.bam $1 --RE $3
echo "partition"
allhic partition all.hic.sorted.dedup.counts_${3}.txt all.hic.sorted.dedup.pairs.txt $2
echo "optimize"
for o in \$(find . -name "all.hic.sorted.dedup.counts_${3}.*g*.txt");
do
echo \$o
allhic optimize \$o all.hic.sorted.dedup.clm
done
echo "build"
allhic build all.hic.sorted.dedup.counts_${3}.${2}g*.tour $1 asm-${2}g.chr.fasta
samtools merge and index
/work/waterhouse_team/apps/ALLHiC/scripts/PreprocessSAMs.pl all.hic.sorted.dedup.bam QMg_NbQ4P_RN.fasta MBOI
Fri Jan 11 09:41:09 2019: PreprocessSAMs.pl: make_bed_around_RE_site.pl QMg_NbQ4P_RN.fasta GATC 500
Fri Jan 11 09:41:09 2019: Reading file QMg_NbQ4P_RN.fasta...
Fri Jan 11 09:42:02 2019: Done! Found 8338823 total instances of motif GATC. Created files:
QMg_NbQ4P_RN.fasta.near_GATC.500.bed
QMg_NbQ4P_RN.fasta.pos_of_GATC.txt
Fri Jan 11 09:42:02 2019: PreprocessSAMs.pl: bedtools intersect -abam all.hic.sorted.dedup.bam -b QMg_NbQ4P_RN.fasta.near_GATC.500.bed > all.hic.sorted.dedup.REduced.bam
Fri Jan 11 17:51:41 2019: PreprocessSAMs.pl: samtools view -F12 all.hic.sorted.dedup.REduced.bam -b -o all.hic.sorted.dedup.REduced.paired_only.bam
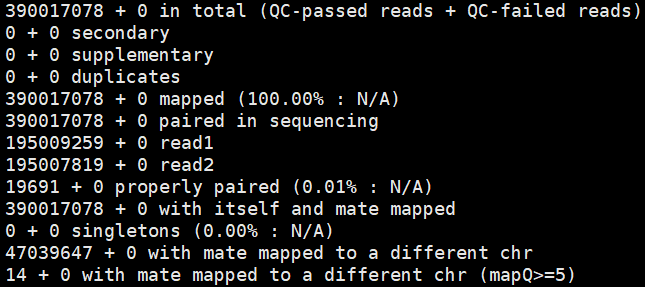
Sat Jan 12 00:58:47 2019: PreprocessSAMs.pl: samtools flagstat all.hic.sorted.dedup.REduced.paired_only.bam > all.hic.sorted.dedup.REduced.paired_only.flagstat
partition
Extract function: calculate an empirical distribution of Hi-C link size based on intra-contig links
CMD: allhic extract sample.clean.bam QMg_NbQ4P_RN.fasta --RE GATC
02:48:59 writeRE | NOTICE RE counts in 1512 contigs (total: 8340335, avg 1 per 332 bp) written to `sample.clean.counts_GATC.txt`
02:48:59 extractContigLinks | NOTICE Parse bamfile `sample.clean.bam`
02:48:59 extractContigLinks | NOTICE Extracted 0 intra-contig link groups (total = 0)
02:48:59 extractContigLinks | NOTICE Extracted 0 inter-contig groups to `sample.clean.clm` (total = 0, maxLinks = 0)
panic: runtime error: index out of range
goroutine 1 [running]:
_/Users/bao/code/allhic.(*LinkDensityModel).countBinDensities(0xc00011a4d0, 0xc000148000, 0x5e8, 0x6a0)
/Users/bao/code/allhic/model.go:149 +0x576
_/Users/bao/code/allhic.(*Extracter).makeModel(0xc00022f740, 0xc000178620, 0x1d)
/Users/bao/code/allhic/extract.go:155 +0x312
_/Users/bao/code/allhic.(*Extracter).Run(0xc00022f740)
/Users/bao/code/allhic/extract.go:139 +0xaf
main.main.func1(0xc0000fcc60, 0xc00001cf00, 0xc0000fcc60)
/Users/bao/code/allhic/cmd/allhic.go:143 +0x17a
github.com/urfave/cli.HandleAction(0x90f200, 0x9da328, 0xc0000fcc60, 0x0, 0xc00001cfc0)
/Users/bao/go/src/github.com/urfave/cli/app.go:501 +0xc8
github.com/urfave/cli.Command.Run(0x9b7b0c, 0x7, 0x0, 0x0, 0x0, 0x0, 0x0, 0x9c5253, 0x23, 0x9d0617, ...)
/Users/bao/go/src/github.com/urfave/cli/command.go:165 +0x459
github.com/urfave/cli.(*App).Run(0xc000140000, 0xc00001c180, 0x6, 0x6, 0x0, 0x0)
/Users/bao/go/src/github.com/urfave/cli/app.go:259 +0x6bb
main.main()
/Users/bao/code/allhic/cmd/allhic.go:400 +0xb31
Partition contigs based on prunning bam file
CMD: allhic partition sample.clean.counts_GATC.txt sample.clean.pairs.txt 38 --minREs 25
02:48:59 ReadCSVLines | NOTICE Parse csvfile `sample.clean.counts_GATC.txt`
02:48:59 readRE | NOTICE Loaded 1512 contig RE lengths for normalization from `sample.clean.counts_GATC.txt`
02:48:59 skipContigsWithFewREs | NOTICE skipContigsWithFewREs with MinREs = 25 (RE = GATC)
Contig #20 (QM4NbP_21) has 4 RE sites -> MARKED SHORT
Contig #32 (QM4NbP_33) has 18 RE sites -> MARKED SHORT
Contig #41 (QM4NbP_42) has 1 RE sites -> MARKED SHORT
Contig #72 (QM4NbP_73) has 2 RE sites -> MARKED SHORT
Contig #75 (QM4NbP_76) has 12 RE sites -> MARKED SHORT
Contig #80 (QM4NbP_81) has 15 RE sites -> MARKED SHORT
Contig #89 (QM4NbP_90) has 23 RE sites -> MARKED SHORT
Contig #113 (QM4NbP_114) has 22 RE sites -> MARKED SHORT
Contig #153 (QM4NbP_154) has 16 RE sites -> MARKED SHORT
Contig #169 (QM4NbP_170) has 6 RE sites -> MARKED SHORT
Contig #177 (QM4NbP_178) has 9 RE sites -> MARKED SHORT
Contig #199 (QM4NbP_200) has 2 RE sites -> MARKED SHORT
Contig #243 (QM4NbP_244) has 1 RE sites -> MARKED SHORT
Contig #282 (QM4NbP_283) has 2 RE sites -> MARKED SHORT
Contig #283 (QM4NbP_284) has 1 RE sites -> MARKED SHORT
Contig #291 (QM4NbP_292) has 1 RE sites -> MARKED SHORT
Contig #293 (QM4NbP_294) has 3 RE sites -> MARKED SHORT
Contig #316 (QM4NbP_317) has 2 RE sites -> MARKED SHORT
Contig #323 (QM4NbP_324) has 24 RE sites -> MARKED SHORT
Contig #343 (QM4NbP_344) has 3 RE sites -> MARKED SHORT
Contig #374 (QM4NbP_375) has 5 RE sites -> MARKED SHORT
Contig #379 (QM4NbP_380) has 10 RE sites -> MARKED SHORT
Contig #384 (QM4NbP_385) has 7 RE sites -> MARKED SHORT
Contig #391 (QM4NbP_392) has 7 RE sites -> MARKED SHORT
Contig #450 (QM4NbP_451) has 7 RE sites -> MARKED SHORT
Contig #470 (QM4NbP_471) has 4 RE sites -> MARKED SHORT
Contig #477 (QM4NbP_478) has 12 RE sites -> MARKED SHORT
Contig #480 (QM4NbP_481) has 23 RE sites -> MARKED SHORT
Contig #488 (QM4NbP_489) has 13 RE sites -> MARKED SHORT
Contig #505 (QM4NbP_506) has 4 RE sites -> MARKED SHORT
02:48:59 skipContigsWithFewREs | NOTICE Marked 30 contigs (avg 8.6 RE sites, len 41439) since they contain too few REs (MinREs = 25)
02:48:59 ReadCSVLines | NOTICE Parse csvfile `sample.clean.pairs.txt`
02:48:59 mustOpen | CRITIC open sample.clean.pairs.txt: no such file or directory
optimize
02:49:02 writeRE | NOTICE RE counts in 1512 contigs (total: 8340335, avg 1 per 332 bp) written to `sample.clean.counts_GATC.txt`
02:49:02 extractContigLinks | NOTICE Parse bamfile `sample.clean.bam`
02:49:02 extractContigLinks | NOTICE Extracted 0 intra-contig link groups (total = 0)
02:49:02 extractContigLinks | NOTICE Extracted 0 inter-contig groups to `sample.clean.clm` (total = 0, maxLinks = 0)
panic: runtime error: index out of range
goroutine 1 [running]:
_/Users/bao/code/allhic.(*LinkDensityModel).countBinDensities(0xc00010e4d0, 0xc000150000, 0x5e8, 0x6a0)
/Users/bao/code/allhic/model.go:149 +0x576
_/Users/bao/code/allhic.(*Extracter).makeModel(0xc000239740, 0xc000182620, 0x1d)
/Users/bao/code/allhic/extract.go:155 +0x312
_/Users/bao/code/allhic.(*Extracter).Run(0xc000239740)
/Users/bao/code/allhic/extract.go:139 +0xaf
main.main.func1(0xc0000f0c60, 0xc00001d000, 0xc0000f0c60)
/Users/bao/code/allhic/cmd/allhic.go:143 +0x17a
github.com/urfave/cli.HandleAction(0x90f200, 0x9da328, 0xc0000f0c60, 0x0, 0xc00001d020)
/Users/bao/go/src/github.com/urfave/cli/app.go:501 +0xc8
github.com/urfave/cli.Command.Run(0x9b7b0c, 0x7, 0x0, 0x0, 0x0, 0x0, 0x0, 0x9c5253, 0x23, 0x9d0617, ...)
/Users/bao/go/src/github.com/urfave/cli/command.go:165 +0x459
github.com/urfave/cli.(*App).Run(0xc000134000, 0xc00001c1e0, 0x6, 0x6, 0x0, 0x0)
/Users/bao/go/src/github.com/urfave/cli/app.go:259 +0x6bb
main.main()
/Users/bao/code/allhic/cmd/allhic.go:400 +0xb31
build
1. tour format to agp ...