Protein structure comparison tools such as SSAP, as used by the Orengo Group in curating CATH.
| Executable DOWNLOADS (for Linux/Mac; chmod them to be executable) |
Docs |
Code |
Extras repo |
 |
cath-cluster Complete-linkage cluster arbitrary data. |
 |
cath-map-clusters Map names from previous clusters to new clusters based on (the overlaps between) their members (which may be specified as regions within a parent sequence). Renumber any clusters with no equivalents. |
 |
cath-resolve-hits Collapse a list of domain matches to your query sequence(s) down to the non-overlapping subset (ie domain architecture) that maximises the sum of the hits' scores. |
 |
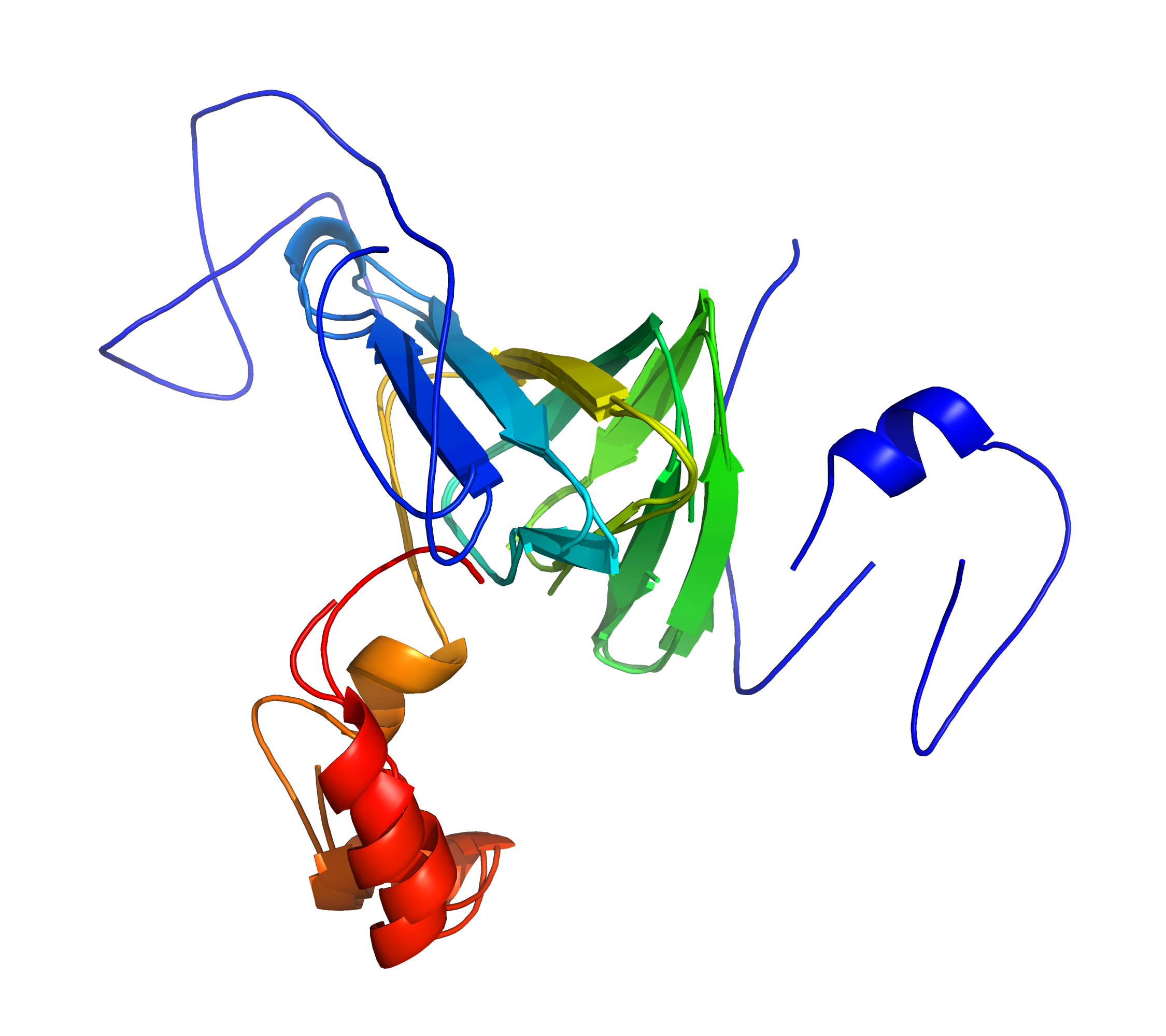
cath-ssap Structurally align a pair of proteins. |
 |
cath-superpose Superpose two or more protein structures using an existing alignment. |
build-testPerform the cath-tools tests (which should all pass, albeit with a few warnings)cath-assign-domainsUse an SVM model on SSAP+PRC data to form a plan for assigning the domains to CATH superfamilies/foldscath-refine-alignIteratively refine an existing alignment by attempting to optimise SSAP scorecath-score-alignScore an existing alignment using structural data
The SSAP algorithm (cath-ssap) was devised by Christine A Orengo and William R Taylor.
Please cite: Protein Structure Alignment, Taylor and Orengo, Journal of Molecular Biology 208, 1-22, PMID: 2769748. (PubMed, Elsevier)
Since then, many people have contributed to this code, most notably:
- Tony E Lewis (2011–…)
- Oliver C Redfern (~2003–2011)
- James E Bray, Ian Sillitoe (~2000–2003)
- Andrew C R Martin (considerable edits around 2001)
cath-ssap typically uses DSSP, either by reading DSSP files or via its own implementation of the DSSP algorithms.
cath-cluster uses Fionn Murtagh's reciprocal-nearest-neighbour algorithm (see Multidimensional clustering algorithms, volume 4 of Compstat Lectures.
Physica-Verlag, Würzburg/ Wien, 1985. ISBN 3-7051-0008-4) as described and refined in Daniel Müllner's Modern hierarchical, agglomerative clustering algorithms (2011, arXiv:1109.2378).
Please tell us about your cath-tools bugs/suggestions here.
If you find this software useful, please spread the word and star the GitHub repo.































































