gao-lab / glue Goto Github PK
View Code? Open in Web Editor NEWGraph-linked unified embedding for single-cell multi-omics data integration
License: MIT License
Graph-linked unified embedding for single-cell multi-omics data integration
License: MIT License












































































































Hi, thank you for this package, it is a great idea for diagonal single cell integration.
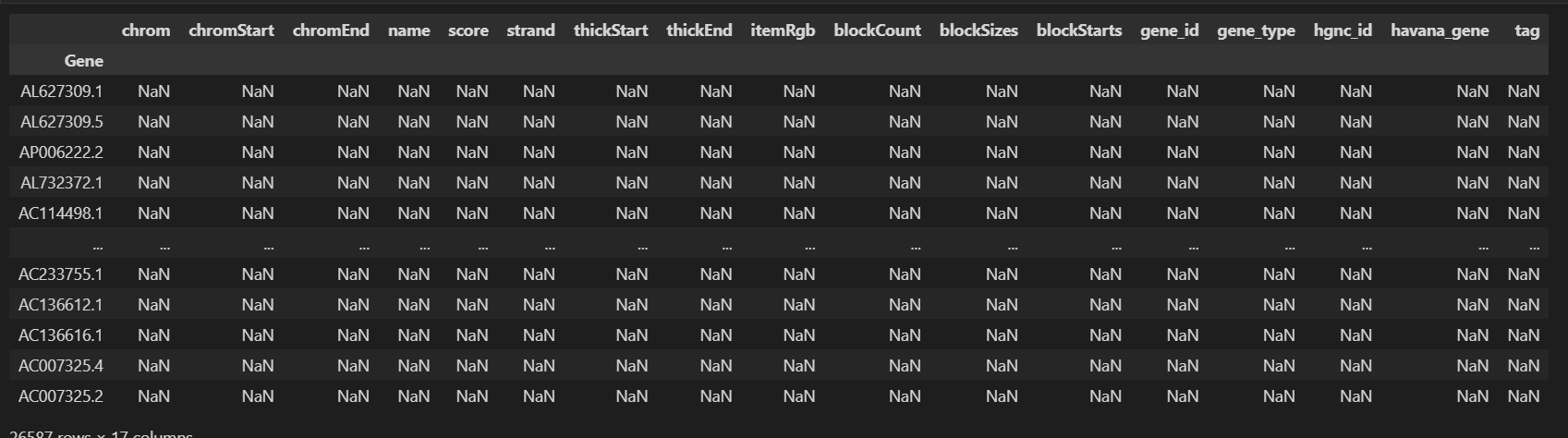
I was going through your tutorial, trying to replicate the pipeline in my data, where I encountered the first error after having NaN values in chromStart and chromEnd fields of my anndata object. I filtered those entries with:
rna = rna[:, pd.notna(rna.var["chromStart"])]After this, I am stumbling upon this other error at the creation of the graph:
graph = scglue.genomics.rna_anchored_prior_graph(rna, atac)
---------------------------------------------------------------------------
ValueError Traceback (most recent call last)
Input In [103], in <module>
----> 1 graph = scglue.genomics.rna_anchored_prior_graph(rna, atac)
2 graph
File ~/conda_envs/csg/lib/python3.9/site-packages/scglue/genomics.py:523, in rna_anchored_prior_graph(rna, gene_region, promoter_len, extend_range, extend_fn, signs, propagate_highly_variable, corrupt_rate, random_state, *others)
520 if set(signs).difference({-1, 1}):
521 raise RuntimeError("``signs`` can only contain {-1, 1}!")
--> 523 rna_bed = Bed(rna.var.assign(name=rna.var_names))
524 other_beds = [Bed(other.var.assign(name=other.var_names)) for other in others]
525 if gene_region == "promoter":
File ~/conda_envs/csg/lib/python3.9/site-packages/scglue/utils.py:479, in ConstrainedDataFrame.__init__(self, *args, **kwargs)
477 df = pd.DataFrame(*args, **kwargs)
478 df = self.rectify(df)
--> 479 self.verify(df)
480 super().__init__(df)
File ~/conda_envs/csg/lib/python3.9/site-packages/scglue/genomics.py:60, in Bed.verify(cls, df)
58 super(Bed, cls).verify(df)
59 if len(df.columns) != len(cls.COLUMNS) or np.any(df.columns != cls.COLUMNS):
---> 60 raise ValueError("Invalid BED format!")
ValueError: Invalid BED format!I have checked the columns and the format of my rna object, the column names that I have are:
rna_test = rna.var.assign(name=rna.var_names)
rna_test.columns
Index(['name', 'highly_variable', 'highly_variable_rank', 'means', 'variances',
'variances_norm', 'chrom', 'chromStart', 'chromEnd', 'name', 'score',
'strand', 'thickStart', 'thickEnd', 'itemRgb', 'blockCount',
'blockSizes', 'blockStarts', 'gene_id', 'gene_type', 'hgnc_id',
'havana_gene', 'tag'],
dtype='object')Could you help me with this?
Thanks!
Thank you for developing such a great tool! I'm working on integrating single cell and spatial transcriptomic data using GLUE. Because they have same var_name(gene name), it is confusing when it comes to prior graph building. I wonder whether I should generate vertice for each dataset, for example, in form of gene_name-dataset , or just build a graph that has gene name as vertices and only contains self-loop?
Hi, I wonder if it is possible to apply glue to match gene expression and protein data, but it seems that we do not have chr information this time, so it would be harder for us to construct the prior graph. Is it possible? Thanks.
Hi, I found that the LSI operation for scATAC-seq data based on dense data has some errors:

If I input an anndata with 400*1000, then I will receive such error:

I tried to fix it by using this operation:

But I meet a new bug here. Could you please help me? Thanks a lot.
I was trying to following the evaluation steps, and have basically installed everything it needs. But I ran into the error of missing data.
Missing input files for rule link_data:
../data/dataset/Chen-2019-FRAGS2RNA.h5ad
Through the link (https://scglue.readthedocs.io/en/latest/data.html) I also didn't see this type of file exist. Do I need to go through all the preproccessing and formatting single-cell data to get that? Btw, the link for the allen_brain.rds is down.
Hi, I have been able to successfully use GLUE to integrate un-paired scRNAseq and scATACseq data.
I was wondering if you have any pointers on using GULE to construct the guidance graph with CyTOF as a third data modality.
Thank you!
Dear GLUE team,
Using scglue version = 0.3.2, I was able to prepare data using the example dataset and the preprocessing notebook successfully. Then, with those RNA/ATAC pre-processed objects, I then get an error (below) while executing the training tutorial (clustering step, post-training at scglue.fit_SCGLUE). It seems linked to differences in the number of edges between the model and data.
I will keep inspecting this error, yet any hint towards a possible solution would be very much appreciated. Thank you.
[INFO] fit_SCGLUE: Pretraining SCGLUE model...
[INFO] check_graph: Checking variable coverage...
[INFO] check_graph: Checking edge attributes...
[INFO] check_graph: Checking self-loops...
[INFO] check_graph: Checking graph symmetry...
[INFO] SCGLUEModel: Setting `graph_batch_size` = 27025
[INFO] SCGLUEModel: Setting `max_epochs` = 186
[INFO] SCGLUEModel: Setting `patience` = 16
[INFO] SCGLUEModel: Setting `reduce_lr_patience` = 8
[INFO] SCGLUETrainer: Using training directory: "glue/pretrain"
[INFO] SCGLUETrainer: [Epoch 10] train={'g_nll': 0.452, 'g_kl': 0.005, 'g_elbo': 0.456, 'x_rna_nll': 0.165, 'x_rna_kl': 0.006, 'x_rna_elbo': 0.172, 'x_atac_nll': 0.04, 'x_atac_kl': 0.0, 'x_atac_elbo': 0.04, 'dsc_loss': 0.692, 'vae_loss': 0.23, 'gen_loss': 0.196}, val={'g_nll': 0.45, 'g_kl': 0.005, 'g_elbo': 0.455, 'x_rna_nll': 0.165, 'x_rna_kl': 0.006, 'x_rna_elbo': 0.171, 'x_atac_nll': 0.04, 'x_atac_kl': 0.0, 'x_atac_elbo': 0.041, 'dsc_loss': 0.693, 'vae_loss': 0.23, 'gen_loss': 0.195}, 10.1s elapsed
[INFO] SCGLUETrainer: [Epoch 20] train={'g_nll': 0.431, 'g_kl': 0.004, 'g_elbo': 0.435, 'x_rna_nll': 0.163, 'x_rna_kl': 0.006, 'x_rna_elbo': 0.17, 'x_atac_nll': 0.04, 'x_atac_kl': 0.0, 'x_atac_elbo': 0.04, 'dsc_loss': 0.693, 'vae_loss': 0.227, 'gen_loss': 0.192}, val={'g_nll': 0.431, 'g_kl': 0.004, 'g_elbo': 0.435, 'x_rna_nll': 0.163, 'x_rna_kl': 0.006, 'x_rna_elbo': 0.169, 'x_atac_nll': 0.04, 'x_atac_kl': 0.0, 'x_atac_elbo': 0.04, 'dsc_loss': 0.694, 'vae_loss': 0.227, 'gen_loss': 0.192}, 10.2s elapsed
[INFO] SCGLUETrainer: [Epoch 30] train={'g_nll': 0.424, 'g_kl': 0.004, 'g_elbo': 0.428, 'x_rna_nll': 0.162, 'x_rna_kl': 0.006, 'x_rna_elbo': 0.168, 'x_atac_nll': 0.04, 'x_atac_kl': 0.0, 'x_atac_elbo': 0.04, 'dsc_loss': 0.693, 'vae_loss': 0.225, 'gen_loss': 0.19}, val={'g_nll': 0.423, 'g_kl': 0.004, 'g_elbo': 0.427, 'x_rna_nll': 0.166, 'x_rna_kl': 0.006, 'x_rna_elbo': 0.172, 'x_atac_nll': 0.04, 'x_atac_kl': 0.0, 'x_atac_elbo': 0.04, 'dsc_loss': 0.693, 'vae_loss': 0.229, 'gen_loss': 0.194}, 9.9s elapsed
Epoch 34: reducing learning rate of group 0 to 2.0000e-04.
Epoch 34: reducing learning rate of group 0 to 2.0000e-04.
[INFO] LRScheduler: Learning rate reduction: step 1
[INFO] SCGLUETrainer: [Epoch 40] train={'g_nll': 0.42, 'g_kl': 0.004, 'g_elbo': 0.424, 'x_rna_nll': 0.161, 'x_rna_kl': 0.006, 'x_rna_elbo': 0.167, 'x_atac_nll': 0.039, 'x_atac_kl': 0.0, 'x_atac_elbo': 0.04, 'dsc_loss': 0.693, 'vae_loss': 0.224, 'gen_loss': 0.189}, val={'g_nll': 0.42, 'g_kl': 0.004, 'g_elbo': 0.424, 'x_rna_nll': 0.164, 'x_rna_kl': 0.006, 'x_rna_elbo': 0.169, 'x_atac_nll': 0.04, 'x_atac_kl': 0.0, 'x_atac_elbo': 0.04, 'dsc_loss': 0.693, 'vae_loss': 0.227, 'gen_loss': 0.192}, 11.6s elapsed
Epoch 45: reducing learning rate of group 0 to 2.0000e-05.
Epoch 45: reducing learning rate of group 0 to 2.0000e-05.
[INFO] LRScheduler: Learning rate reduction: step 2
[INFO] SCGLUETrainer: [Epoch 50] train={'g_nll': 0.42, 'g_kl': 0.004, 'g_elbo': 0.424, 'x_rna_nll': 0.161, 'x_rna_kl': 0.006, 'x_rna_elbo': 0.167, 'x_atac_nll': 0.039, 'x_atac_kl': 0.0, 'x_atac_elbo': 0.04, 'dsc_loss': 0.693, 'vae_loss': 0.223, 'gen_loss': 0.189}, val={'g_nll': 0.42, 'g_kl': 0.004, 'g_elbo': 0.423, 'x_rna_nll': 0.161, 'x_rna_kl': 0.006, 'x_rna_elbo': 0.167, 'x_atac_nll': 0.04, 'x_atac_kl': 0.0, 'x_atac_elbo': 0.04, 'dsc_loss': 0.693, 'vae_loss': 0.225, 'gen_loss': 0.19}, 10.6s elapsed
2022-10-04 14:44:51,492 ignite.handlers.early_stopping.EarlyStopping INFO: EarlyStopping: Stop training
[INFO] EarlyStopping: Restoring checkpoint "52"...
[INFO] fit_SCGLUE: Estimating balancing weight...
[INFO] estimate_balancing_weight: Clustering cells...
---------------------------------------------------------------------------
BaseException Traceback (most recent call last)
Cell In [14], line 1
----> 1 glue = scglue.models.fit_SCGLUE(
2 {"rna": rna, "atac": atac}, guidance_hvf,
3 fit_kws={"directory": "glue"}
4 )
File ~/miniconda3/envs/scglue/lib/python3.8/site-packages/scglue/models/__init__.py:221, in fit_SCGLUE(adatas, graph, model, init_kws, compile_kws, fit_kws, balance_kws)
219 else:
220 use_batch = None
--> 221 estimate_balancing_weight(
222 *adatas.values(), use_rep=f"X_{config.TMP_PREFIX}", use_batch=use_batch,
223 key_added="balancing_weight", **balance_kws
224 )
225 for adata in adatas.values():
226 adata.uns[config.ANNDATA_KEY]["use_dsc_weight"] = "balancing_weight"
File ~/miniconda3/envs/scglue/lib/python3.8/site-packages/scglue/data.py:469, in estimate_balancing_weight(use_rep, use_batch, resolution, cutoff, power, key_added, *adatas)
464 for adata_ in adatas_:
465 sc.pp.neighbors(
466 adata_, n_pcs=adata_.obsm[use_rep].shape[1],
467 use_rep=use_rep, metric="cosine"
468 )
--> 469 sc.tl.leiden(adata_, resolution=resolution)
471 leidens = [
472 aggregate_obs(
473 adata, by="leiden", X_agg=None,
474 obs_agg={"n": "sum"}, obsm_agg={use_rep: "mean"}
475 ) for adata in adatas_
476 ]
477 us = [normalize(leiden.obsm[use_rep], norm="l2") for leiden in leidens]
File ~/miniconda3/envs/scglue/lib/python3.8/site-packages/scanpy/tools/_leiden.py:144, in leiden(adata, resolution, restrict_to, random_state, key_added, adjacency, directed, use_weights, n_iterations, partition_type, neighbors_key, obsp, copy, **partition_kwargs)
142 partition_kwargs['resolution_parameter'] = resolution
143 # clustering proper
--> 144 part = leidenalg.find_partition(g, partition_type, **partition_kwargs)
145 # store output into adata.obs
146 groups = np.array(part.membership)
File ~/miniconda3/envs/scglue/lib/python3.8/site-packages/leidenalg/functions.py:81, in find_partition(graph, partition_type, initial_membership, weights, n_iterations, max_comm_size, seed, **kwargs)
79 if not weights is None:
80 kwargs['weights'] = weights
---> 81 partition = partition_type(graph,
82 initial_membership=initial_membership,
83 **kwargs)
84 optimiser = Optimiser()
86 optimiser.max_comm_size = max_comm_size
File ~/miniconda3/envs/scglue/lib/python3.8/site-packages/leidenalg/VertexPartition.py:840, in RBConfigurationVertexPartition.__init__(self, graph, initial_membership, weights, resolution_parameter)
836 else:
837 # Make sure it is a list
838 weights = list(weights)
--> 840 self._partition = _c_leiden._new_RBConfigurationVertexPartition(pygraph_t,
841 initial_membership, weights, resolution_parameter)
842 self._update_internal_membership()
BaseException: Could not construct partition: Weight vector not the same size as the number of edges.
See
Line 609 in e20518b
integration_consistency makes this requirement. I am wondering why this is the case? I have floating point data I am trying to integrate.when I run Like the following,
glue = scglue.models.fit_SCGLUE(
{"rna": rna, "atac": atac}, graph,
fit_kws={"directory": "glue2"}
)
dx = scglue.models.integration_consistency(
glue, {"rna": rna, "atac": atac}, graph,
count_layers={"rna": "counts","atac": "counts"}
)
dx
_ = sns.lineplot(x="n_meta", y="consistency", data=dx).axhline(y=0.05, c="darkred", ls="--")
rna.obsm["X_glue"] = glue.encode_data("rna", rna)
atac.obsm["X_glue"] = glue.encode_data("atac", atac)
combined = anndata.concat([rna, atac])I get this combined.
AnnData object with n_obs × n_vars = 592831 × 0
obs: 'sample', 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'patient', 'tissue', 'balancing_weight'
uns: 'neighbors', 'umap'
obsm: 'X_umap', 'X_glue'
layers: 'counts'
obsp: 'distances', 'connectivities'
I can't understand it !!!!
Dear developers,
Thank you for sharing your code. I want to take this opportunity to ask whether we can use your Glue program to integrate simple single-cell transcriptome with general proteomics and metabonomics. This is very important to me. Thank you for your answer.
Best wishes,
zjz
I use the scglue.data.transfer_labels to transfer the labels from RNA to atac. Does scglue have similar functions to get the predicted.labels that are used in singac to Evaluate the prediction and Confidence score of ATAC cells? Will scglue retain all cells in ATAC?
Hi, sorry to disturb you again. After reviewing your process related to data preprocessing, I am wondering if you utilize the highly variable genes selection process in these steps:
print('Preprocessing GEX...')
gex_prep = utils.GEXPreprocessing(n_comps=100, n_genes=n_genes, merge_adt=omics == "cite")
gex_prep.fit_transform(gex)
if omics == "cite":
print('Preprocessing ADT...')
other_prep = utils.ADTPreprocessing(n_comps=100)
elif omics == "multiome":
print('Preprocessing ATAC...')
other_prep = utils.ATACPreprocessing(n_comps=100)
Could you please share your ideas about hvg selection with me? If I did hvg selection, will the matching become more accuracy? Thanks a lot.
Hi, I finally generated the model of glue, but met a new problem:
In this code:

I meet this problem:

I think it may be caused by the sparse data type of counts data:

Could you please help me? Thanks a lot.
Hi,
Thanks for the register function - works great!
I'm wondering why the batch effect mechanism requires that the datasets have identical batches - are batches passed in during training during the training i.e only one batch used for every mini-batch? Is there a reason for this?
Dear Dr. Cao, sorry to disturb you. When I intend to run CLUE based on the open source all datasets provided by the organizers of neurips competition, I find an error:

Even if I run this method before training, I still receive this error for clue.fitting step:

In addition, I think I cannot find these two datasets in the open codes part of this page: https://github.com/openproblems-bio/neurips2021_multimodal_topmethods/tree/main/src/match_modality/methods/clue
including the file prep.pickle and "hyperparams.yaml. I think without these files I cannot utilize the best combination of parameters.


Could you please help me? Thanks a lot.
I have scATAC and scRNA from multiple different samples, can I directly integrate them with glue?
Hi Dr. Cao, I come back with a confusing problem after I re-run the code related to clue, which is published by the single cell problems organizers.

I think this code can yield cell embeddings with dim=50 if I set latent dim as 50, however, even if I set such things, I still get dim=100 data, which is the same as the dimensions of network input, for this model.

Is there anything wrong with my understanding? Thanks a lot.
Thanks for developing a nice tool! I used glue to integrate scRNA-seq and scATAC-seq datasets from multiple samples. I want to get clustering results covering both modalities in the umap plot.
What can I do to get this information? Can I do clustering on embedding space?
Can I come up with batch-corrected data for clustering?
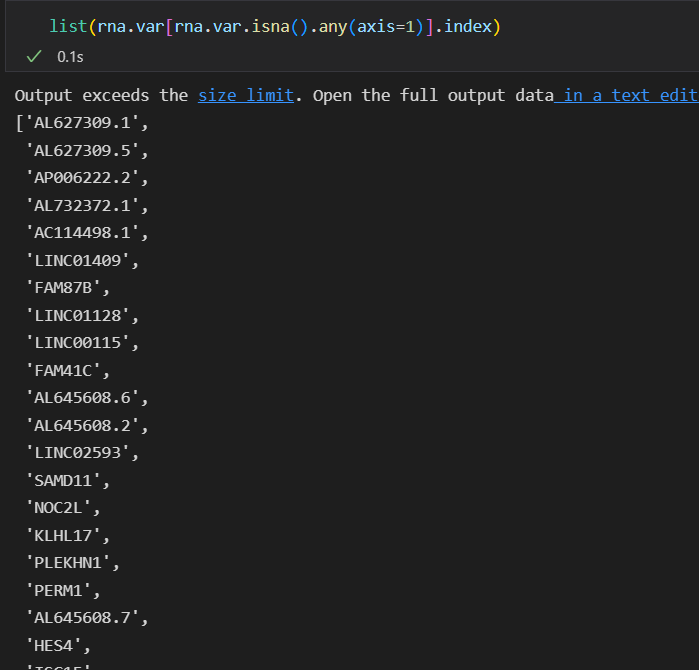
When I run scglue.data.get_gene_annotation to obtain genomic coordinates, occurs this problem.
https://raw.githubusercontent.com/jisnoalia/image/master/img/202205201031888.png
len( list(rna.var[~rna.var.isna().any(axis=1)].index))
6236
len( list(rna.var[rna.var.isna().any(axis=1)].index))
20351https://raw.githubusercontent.com/jisnoalia/image/master/img/202205201039666.png
But I can find the gene like FAM87B in gtf file.I don't know if this problem affects the analysis.
ValueError: Expected value argument (Tensor of shape (128, 2000)) to be within the support (IntegerGreaterThan(lower_bound=0)) of the distribution NegativeBinomial(total_count: torch.Size([128, 2000]), logits: torch.Size([128, 2000])), but found invalid values:

An error was reported when I ran tutorial code scglue.genomics.rna_anchored_prior_graph(rna, atac)
I have installed sortBed
graph = scglue.genomics.rna_anchored_prior_graph(rna, atac)
NotImplementedError Traceback (most recent call last)
/tmp/ipykernel_176444/2387691421.py in <module>
1 import pybedtools
----> 2 graph = scglue.genomics.rna_anchored_prior_graph(rna, atac)
3 graph
~/miniconda3/envs/pytorch/lib/python3.7/site-packages/scglue/genomics.py in rna_anchored_prior_graph(rna, gene_region, promoter_len, extend_range, extend_fn, signs, propagate_highly_variable, corrupt_rate, random_state, *others)
534 "dist": abs(d), "weight": extend_fn(abs(d)), "sign": s
535 }
--> 536 ) for other_bed, sign in zip(other_beds, signs)]
537 graph = compose_multigraph(*graphs)
538
~/miniconda3/envs/pytorch/lib/python3.7/site-packages/scglue/genomics.py in <listcomp>(.0)
534 "dist": abs(d), "weight": extend_fn(abs(d)), "sign": s
535 }
--> 536 ) for other_bed, sign in zip(other_beds, signs)]
537 graph = compose_multigraph(*graphs)
538
~/miniconda3/envs/pytorch/lib/python3.7/site-packages/scglue/genomics.py in window_graph(left, right, window_size, left_sorted, right_sorted, attr_fn)
409 left = pybedtools.BedTool(left)
410 if not left_sorted:
--> 411 left = left.sort(stream=True)
412 left = iter(left) # Resumable iterator
413 if isinstance(right, Bed):
~/miniconda3/envs/pytorch/lib/python3.7/site-packages/pybedtools/bedtool.py in decorated(self, *args, **kwargs)
921 # this calls the actual method in the first place; *result* is
922 # whatever you get back
--> 923 result = method(self, *args, **kwargs)
924
925 # add appropriate tags
~/miniconda3/envs/pytorch/lib/python3.7/site-packages/pybedtools/bedtool.py in not_implemented_func(*args, **kwargs)
242
243 def not_implemented_func(*args, **kwargs):
--> 244 raise NotImplementedError(help_str)
245
246 return not_implemented_func
NotImplementedError: "sortBed" does not appear to be installed or on the path, so this method is disabled. Please install a more recent version of BEDTools and re-import to use this method.

Hi,I'm using GLUE integrated my data,but when i running scglue.genomics.rna_anchored_prior_graph(rna, atac). I get follow error:
ValueError Traceback (most recent call last)
/tmp/ipykernel_374527/446942525.py in <module>
----> 1 graph = scglue.genomics.rna_anchored_prior_graph(rna, atac)
~/miniconda3/envs/pyscenic/lib/python3.7/site-packages/scglue/genomics.py in rna_anchored_prior_graph(rna, gene_region, promoter_len, extend_range, extend_fn, signs, propagate_highly_variable, corrupt_rate, random_state, *others)
522
523 rna_bed = Bed(rna.var.assign(name=rna.var_names))
--> 524 other_beds = [Bed(other.var.assign(name=other.var_names)) for other in others]
525 if gene_region == "promoter":
526 rna_bed = rna_bed.strand_specific_start_site().expand(promoter_len, 0)
~/miniconda3/envs/pyscenic/lib/python3.7/site-packages/scglue/genomics.py in <listcomp>(.0)
522
523 rna_bed = Bed(rna.var.assign(name=rna.var_names))
--> 524 other_beds = [Bed(other.var.assign(name=other.var_names)) for other in others]
525 if gene_region == "promoter":
526 rna_bed = rna_bed.strand_specific_start_site().expand(promoter_len, 0)
~/miniconda3/envs/pyscenic/lib/python3.7/site-packages/scglue/utils.py in __init__(self, *args, **kwargs)
491 def __init__(self, *args, **kwargs) -> None:
492 df = pd.DataFrame(*args, **kwargs)
--> 493 df = self.rectify(df)
494 self.verify(df)
495 super().__init__(df)
~/miniconda3/envs/pyscenic/lib/python3.7/site-packages/scglue/genomics.py in rectify(cls, df)
45 if item in df:
46 if item in ("chromStart", "chromEnd"):
---> 47 df[item] = df[item].astype(int)
48 else:
49 df[item] = df[item].astype(str)
~/miniconda3/envs/pyscenic/lib/python3.7/site-packages/pandas/core/generic.py in astype(self, dtype, copy, errors)
5813 else:
5814 # else, only a single dtype is given
-> 5815 new_data = self._mgr.astype(dtype=dtype, copy=copy, errors=errors)
5816 return self._constructor(new_data).__finalize__(self, method="astype")
5817
~/miniconda3/envs/pyscenic/lib/python3.7/site-packages/pandas/core/internals/managers.py in astype(self, dtype, copy, errors)
416
417 def astype(self: T, dtype, copy: bool = False, errors: str = "raise") -> T:
--> 418 return self.apply("astype", dtype=dtype, copy=copy, errors=errors)
419
420 def convert(
~/miniconda3/envs/pyscenic/lib/python3.7/site-packages/pandas/core/internals/managers.py in apply(self, f, align_keys, ignore_failures, **kwargs)
325 applied = b.apply(f, **kwargs)
326 else:
--> 327 applied = getattr(b, f)(**kwargs)
328 except (TypeError, NotImplementedError):
329 if not ignore_failures:
~/miniconda3/envs/pyscenic/lib/python3.7/site-packages/pandas/core/internals/blocks.py in astype(self, dtype, copy, errors)
589 values = self.values
590
--> 591 new_values = astype_array_safe(values, dtype, copy=copy, errors=errors)
592
593 new_values = maybe_coerce_values(new_values)
~/miniconda3/envs/pyscenic/lib/python3.7/site-packages/pandas/core/dtypes/cast.py in astype_array_safe(values, dtype, copy, errors)
1307
1308 try:
-> 1309 new_values = astype_array(values, dtype, copy=copy)
1310 except (ValueError, TypeError):
1311 # e.g. astype_nansafe can fail on object-dtype of strings
~/miniconda3/envs/pyscenic/lib/python3.7/site-packages/pandas/core/dtypes/cast.py in astype_array(values, dtype, copy)
1255
1256 else:
-> 1257 values = astype_nansafe(values, dtype, copy=copy)
1258
1259 # in pandas we don't store numpy str dtypes, so convert to object
~/miniconda3/envs/pyscenic/lib/python3.7/site-packages/pandas/core/dtypes/cast.py in astype_nansafe(arr, dtype, copy, skipna)
1172 # work around NumPy brokenness, #1987
1173 if np.issubdtype(dtype.type, np.integer):
-> 1174 return lib.astype_intsafe(arr, dtype)
1175
1176 # if we have a datetime/timedelta array of objects
~/miniconda3/envs/pyscenic/lib/python3.7/site-packages/pandas/_libs/lib.pyx in pandas._libs.lib.astype_intsafe()
ValueError: invalid literal for int() with base 10: 'ctg150'
How can i fix it?
https://www.dropbox.com/s/dl/kqsy9tvsklbu7c4/allen_brain.rds cann't use in 'GLUE/data/download/10x-ATAC-Brain5k'
Hi, I looked at the recoding of your open problem presentation and I was wondering 1) if there's a careful feature selection for either the ATACseq data and the RNAseq data? 20 how many scATACseq peaks and scRNAseq genes you selected for the model training? Thanks!
Hi,your work about GLUE is very good,but i have a few question.
Your article mentioned that with the use of omic data ,we can build GRN through our method.But i can not find the code.
Will you be so kind to provide the code with me?
Yours,
Dan
Dear Dr. Cao,
Sorry to disturb you again. I take a look at the code you used in the NeuriPS competition and I wonder why you choose to reduce the input dimensions from 100 to 50 as latent space? It seems that you only reduced half of the original space. From my perspective, this approach may not help to improve the performance of the final matching.
Thanks a lot!
Hi there, thanks for the very helpful package, GLUE!
I am integrating single-cell RNA-seq and ATAC-seq data where one cluster is missing in one dataset, so the sizes of the two datasets are different. When running GLUE, I get the following error, could you help me with this? Is the error happening because of the size difference between the number of cells in RNA-seq (707 cells) and ATAC-seq (1047 cells) matrices?
I followed the tutorials, but at this step I get an error:
glue = scglue.models.fit_SCGLUE(
{"rna": rna, "atac": atac}, graph,
fit_kws={"directory": "glue"}
)
[INFO] fit_SCGLUE: Pretraining SCGLUE model...
[INFO] autodevice: Using GPU 1 as computation device.
[INFO] SCGLUEModel: Setting graph_batch_size = 11614
[INFO] SCGLUEModel: Setting max_epochs = 1947
[INFO] SCGLUEModel: Setting patience = 163
[INFO] SCGLUEModel: Setting reduce_lr_patience = 82
[INFO] SCGLUETrainer: Using training directory: "glue/pretrain"
ValueError Traceback (most recent call last)
in
1 glue = scglue.models.fit_SCGLUE(
2 {"rna": rna, "atac": atac}, graph,
----> 3 fit_kws={"directory": "glue"}
4 )
~/.local/lib/python3.6/site-packages/scglue/models/init.py in fit_SCGLUE(adatas, graph, model, init_kws, compile_kws, fit_kws, balance_kws)
89 pretrain = model(adatas, sorted(graph.nodes), **pretrain_init_kws)
90 pretrain.compile(**compile_kws)
---> 91 pretrain.fit(adatas, graph, **pretrain_fit_kws)
92 if "directory" in pretrain_fit_kws:
93 pretrain.save(os.path.join(pretrain_fit_kws["directory"], "pretrain.dill"))
~/.local/lib/python3.6/site-packages/scglue/models/scglue.py in fit(self, adatas, graph, edge_weight, edge_sign, neg_samples, val_split, data_batch_size, graph_batch_size, align_burnin, safe_burnin, max_epochs, patience, reduce_lr_patience, wait_n_lrs, directory)
1516 reduce_lr_patience=reduce_lr_patience, wait_n_lrs=wait_n_lrs,
1517 random_seed=self.random_seed,
-> 1518 directory=directory
1519 )
1520
~/.local/lib/python3.6/site-packages/scglue/models/base.py in fit(self, *args, **kwargs)
332 Subclasses may override arguments for API definition.
333 """
--> 334 self.trainer.fit(*args, **kwargs)
335
336 def get_losses(self, *args, **kwargs) -> Mapping[str, float]:
~/.local/lib/python3.6/site-packages/scglue/models/glue.py in fit(self, data, graph, val_split, data_batch_size, graph_batch_size, align_burnin, safe_burnin, max_epochs, patience, reduce_lr_patience, wait_n_lrs, random_seed, directory, plugins)
609 train_loader, val_loader=val_loader,
610 max_epochs=max_epochs, random_seed=random_seed,
--> 611 directory=directory, plugins=plugins
612 )
613 finally:
~/.local/lib/python3.6/site-packages/scglue/models/base.py in fit(self, train_loader, val_loader, max_epochs, random_seed, directory, plugins)
198 # Start engines
199 torch.manual_seed(random_seed)
--> 200 train_engine.run(train_loader, max_epochs=max_epochs)
201
202 torch.cuda.empty_cache() # Works even if GPU is unavailable
~/.local/lib/python3.6/site-packages/ignite/engine/engine.py in run(self, data, max_epochs, epoch_length, seed)
702
703 self.state.dataloader = data
--> 704 return self._internal_run()
705
706 @staticmethod
~/.local/lib/python3.6/site-packages/ignite/engine/engine.py in _internal_run(self)
781 self._dataloader_iter = None
782 self.logger.error(f"Engine run is terminating due to exception: {e}")
--> 783 self._handle_exception(e)
784
785 self._dataloader_iter = None
~/.local/lib/python3.6/site-packages/ignite/engine/engine.py in _handle_exception(self, e)
462 def _handle_exception(self, e: BaseException) -> None:
463 if Events.EXCEPTION_RAISED in self._event_handlers:
--> 464 self._fire_event(Events.EXCEPTION_RAISED, e)
465 else:
466 raise e
~/.local/lib/python3.6/site-packages/ignite/engine/engine.py in _fire_event(self, event_name, *event_args, **event_kwargs)
419 kwargs.update(event_kwargs)
420 first, others = ((args[0],), args[1:]) if (args and args[0] == self) else ((), args)
--> 421 func(*first, *(event_args + others), **kwargs)
422
423 def fire_event(self, event_name: Any) -> None:
~/.local/lib/python3.6/site-packages/scglue/models/base.py in _handle_exception(engine, e)
161 engine.terminate()
162 else:
--> 163 raise e
164
165 # Compute metrics
~/.local/lib/python3.6/site-packages/ignite/engine/engine.py in _internal_run(self)
751 self._setup_engine()
752
--> 753 time_taken = self._run_once_on_dataset()
754 # time is available for handlers but must be update after fire
755 self.state.times[Events.EPOCH_COMPLETED.name] = time_taken
~/.local/lib/python3.6/site-packages/ignite/engine/engine.py in _run_once_on_dataset(self)
852 except Exception as e:
853 self.logger.error(f"Current run is terminating due to exception: {e}")
--> 854 self._handle_exception(e)
855
856 return time.time() - start_time
~/.local/lib/python3.6/site-packages/ignite/engine/engine.py in _handle_exception(self, e)
462 def _handle_exception(self, e: BaseException) -> None:
463 if Events.EXCEPTION_RAISED in self._event_handlers:
--> 464 self._fire_event(Events.EXCEPTION_RAISED, e)
465 else:
466 raise e
~/.local/lib/python3.6/site-packages/ignite/engine/engine.py in _fire_event(self, event_name, *event_args, **event_kwargs)
419 kwargs.update(event_kwargs)
420 first, others = ((args[0],), args[1:]) if (args and args[0] == self) else ((), args)
--> 421 func(*first, *(event_args + others), **kwargs)
422
423 def fire_event(self, event_name: Any) -> None:
~/.local/lib/python3.6/site-packages/scglue/models/base.py in _handle_exception(engine, e)
161 engine.terminate()
162 else:
--> 163 raise e
164
165 # Compute metrics
~/.local/lib/python3.6/site-packages/ignite/engine/engine.py in _run_once_on_dataset(self)
838 self.state.iteration += 1
839 self._fire_event(Events.ITERATION_STARTED)
--> 840 self.state.output = self._process_function(self, self.state.batch)
841 self._fire_event(Events.ITERATION_COMPLETED)
842
~/.local/lib/python3.6/site-packages/scglue/models/scglue.py in train_step(self, engine, data)
726
727 # Generator step
--> 728 losses = self.compute_losses(data, epoch)
729 self.net.zero_grad(set_to_none=True)
730 losses["gen_loss"].backward()
~/.local/lib/python3.6/site-packages/scglue/models/scglue.py in compute_losses(self, data, epoch, dsc_only)
676 usamp[k], vsamp[getattr(net, f"{k}_idx")], xbch[k], l[k]
677 ).log_prob(x[k]).mean()
--> 678 for k in net.keys
679 }
680 x_kl = {
~/.local/lib/python3.6/site-packages/scglue/models/scglue.py in (.0)
676 usamp[k], vsamp[getattr(net, f"{k}_idx")], xbch[k], l[k]
677 ).log_prob(x[k]).mean()
--> 678 for k in net.keys
679 }
680 x_kl = {
~/.local/lib/python3.6/site-packages/torch/distributions/negative_binomial.py in log_prob(self, value)
90 def log_prob(self, value):
91 if self._validate_args:
---> 92 self._validate_sample(value)
93
94 log_unnormalized_prob = (self.total_count * F.logsigmoid(-self.logits) +
~/.local/lib/python3.6/site-packages/torch/distributions/distribution.py in _validate_sample(self, value)
275 assert support is not None
276 if not support.check(value).all():
--> 277 raise ValueError('The value argument must be within the support')
278
279 def _get_checked_instance(self, cls, _instance=None):
ValueError: The value argument must be within the support
> rna
AnnData object with n_obs × n_vars = 707 × 10856
obs: 'n_genes', 'domain'
var: 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std', 'chrom', 'chromStart', 'chromEnd', 'name', 'score', 'strand', 'thickStart', 'thickEnd', 'itemRgb', 'blockCount', 'blockSizes', 'blockStarts', 'gene_id', 'gene_type', 'hgnc_id', 'havana_gene', 'tag'
uns: 'hvg', 'log1p', 'pca', 'neighbors', 'umap', '__scglue__'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts'
obsp: 'distances', 'connectivities'
> atac
AnnData object with n_obs × n_vars = 1047 × 136771
obs: 'domain'
var: 'chrom', 'chromStart', 'chromEnd', 'highly_variable'
uns: 'neighbors', 'umap', '__scglue__'
obsm: 'X_lsi', 'X_umap'
obsp: 'distances', 'connectivities'
Thanks!
Hi
I am following pipeline from the GLUE publication to get the regulatory inference.
In that attempt, I am at /GLUE/tree/master/experiments/RegInf)/s03_peak_gene_validation.py
However I get following error
ValueError Traceback (most recent call last)
/tmp/ipykernel_3936564/4102559450.py in
1 corr = biadjacency_matrix(
2 utils.metacell_corr(
----> 3 rna, atac, "X_UMAP", n_meta=200, skeleton=dist_graph, method="spr"
4 ), genes.index, peaks.index, weight="corr", dtype=np.float32
5 )
~/utils.py in metacell_corr(rna, atac, use_rep, n_meta, skeleton, method)
26 def metacell_corr(rna, atac, use_rep, n_meta=200, skeleton=None, method="spr"):
27 print("Clustering metacells...")
---> 28 rna_agg, atac_agg = get_metacells_paired(rna, atac, use_rep, n_meta=n_meta)
29 print("Computing correlation...")
30 return _metacell_corr(rna_agg, atac_agg, skeleton=skeleton, method=method)
~/utils.py in get_metacells_paired(rna, atac, use_rep, n_meta)
15 kmeans.train(rna.obsm[use_rep])
16 _, rna.obs["metacell"] = kmeans.index.search(rna.obsm[use_rep], 1)
---> 17 atac.obs["metacell"] = rna.obs["metacell"].to_numpy()
18 rna_agg = scglue.data.aggregate_obs(rna, "metacell")
19 atac_agg = scglue.data.aggregate_obs(atac, "metacell")
~/miniconda3/envs/mypython3/lib/python3.7/site-packages/pandas/core/frame.py in setitem(self, key, value)
3610 else:
3611 # set column
-> 3612 self._set_item(key, value)
3613
3614 def _setitem_slice(self, key: slice, value):
~/miniconda3/envs/mypython3/lib/python3.7/site-packages/pandas/core/frame.py in _set_item(self, key, value)
3782 ensure homogeneity.
3783 """
-> 3784 value = self._sanitize_column(value)
3785
3786 if (
~/miniconda3/envs/mypython3/lib/python3.7/site-packages/pandas/core/frame.py in _sanitize_column(self, value)
4507
4508 if is_list_like(value):
-> 4509 com.require_length_match(value, self.index)
4510 return sanitize_array(value, self.index, copy=True, allow_2d=True)
4511
~/miniconda3/envs/mypython3/lib/python3.7/site-packages/pandas/core/common.py in require_length_match(data, index)
530 if len(data) != len(index):
531 raise ValueError(
--> 532 "Length of values "
533 f"({len(data)}) "
534 "does not match length of index "
Does 'rna' and 'atac' use_rep need to be the same size?
Can someone please explain the error
Hi , thanks for the very helpful package!
I have used the method in the #22 (comment), but it didn't solve my question.Could you give me a help?Thanks!
BEST!
XUE


Hi,when i run the scglue.models.fit_SCGLUE,
glue = scglue.models.fit_SCGLUE( {"rna": rna, "atac": atac}, graph, fit_kws={"directory": "glue"} )
my RNA and ATAC are transferred from Seurat.
it shows this error :
use_rep dimensionality be equal or larger than latent_dim.graph_batch_size = 11613max_epochs = 48patience = 4reduce_lr_patience = 2ValueError Traceback (most recent call last)
Input In [58], in <cell line: 1>()
----> 1 glue = scglue.models.fit_SCGLUE(
2 {"rna": rna, "atac": atac}, graph,
3 fit_kws={"directory": "glue"}
4 )
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/scglue/models/init.py:91, in fit_SCGLUE(adatas, graph, model, init_kws, compile_kws, fit_kws, balance_kws)
89 pretrain = model(adatas, sorted(graph.nodes), **pretrain_init_kws)
90 pretrain.compile(**compile_kws)
---> 91 pretrain.fit(adatas, graph, **pretrain_fit_kws)
92 if "directory" in pretrain_fit_kws:
93 pretrain.save(os.path.join(pretrain_fit_kws["directory"], "pretrain.dill"))
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/scglue/models/scglue.py:1511, in SCGLUEModel.fit(self, adatas, graph, edge_weight, edge_sign, neg_samples, val_split, data_batch_size, graph_batch_size, align_burnin, safe_burnin, max_epochs, patience, reduce_lr_patience, wait_n_lrs, directory)
1508 if self.trainer.freeze_u:
1509 self.logger.info("Cell embeddings are frozen")
-> 1511 super().fit(
1512 data, graph, val_split=val_split,
1513 data_batch_size=data_batch_size, graph_batch_size=graph_batch_size,
1514 align_burnin=align_burnin, safe_burnin=safe_burnin,
1515 max_epochs=max_epochs, patience=patience,
1516 reduce_lr_patience=reduce_lr_patience, wait_n_lrs=wait_n_lrs,
1517 random_seed=self.random_seed,
1518 directory=directory
1519 )
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/scglue/models/base.py:334, in Model.fit(self, *args, **kwargs)
319 def fit(self, *args, **kwargs) -> None:
320 r"""
321 Alias of .trainer.fit.
322
(...)
332 Subclasses may override arguments for API definition.
333 """
--> 334 self.trainer.fit(*args, **kwargs)
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/scglue/models/glue.py:608, in GLUETrainer.fit(self, data, graph, val_split, data_batch_size, graph_batch_size, align_burnin, safe_burnin, max_epochs, patience, reduce_lr_patience, wait_n_lrs, random_seed, directory, plugins)
606 plugins = default_plugins + (plugins or [])
607 try:
--> 608 super().fit(
609 train_loader, val_loader=val_loader,
610 max_epochs=max_epochs, random_seed=random_seed,
611 directory=directory, plugins=plugins
612 )
613 finally:
614 data.clean()
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/scglue/models/base.py:200, in Trainer.fit(self, train_loader, val_loader, max_epochs, random_seed, directory, plugins)
198 # Start engines
199 torch.manual_seed(random_seed)
--> 200 train_engine.run(train_loader, max_epochs=max_epochs)
202 torch.cuda.empty_cache()
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/ignite/engine/engine.py:704, in Engine.run(self, data, max_epochs, epoch_length, seed)
701 raise ValueError("epoch_length should be provided if data is None")
703 self.state.dataloader = data
--> 704 return self._internal_run()
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/ignite/engine/engine.py:783, in Engine._internal_run(self)
781 self._dataloader_iter = None
782 self.logger.error(f"Engine run is terminating due to exception: {e}")
--> 783 self._handle_exception(e)
785 self._dataloader_iter = None
786 return self.state
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/ignite/engine/engine.py:464, in Engine._handle_exception(self, e)
462 def _handle_exception(self, e: BaseException) -> None:
463 if Events.EXCEPTION_RAISED in self._event_handlers:
--> 464 self._fire_event(Events.EXCEPTION_RAISED, e)
465 else:
466 raise e
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/ignite/engine/engine.py:421, in Engine._fire_event(self, event_name, *event_args, **event_kwargs)
419 kwargs.update(event_kwargs)
420 first, others = ((args[0],), args[1:]) if (args and args[0] == self) else ((), args)
--> 421 func(*first, *(event_args + others), **kwargs)
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/scglue/models/base.py:163, in Trainer.fit.._handle_exception(engine, e)
161 engine.terminate()
162 else:
--> 163 raise e
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/ignite/engine/engine.py:753, in Engine._internal_run(self)
750 if self._dataloader_iter is None:
751 self._setup_engine()
--> 753 time_taken = self._run_once_on_dataset()
754 # time is available for handlers but must be update after fire
755 self.state.times[Events.EPOCH_COMPLETED.name] = time_taken
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/ignite/engine/engine.py:854, in Engine._run_once_on_dataset(self)
852 except Exception as e:
853 self.logger.error(f"Current run is terminating due to exception: {e}")
--> 854 self._handle_exception(e)
856 return time.time() - start_time
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/ignite/engine/engine.py:464, in Engine._handle_exception(self, e)
462 def _handle_exception(self, e: BaseException) -> None:
463 if Events.EXCEPTION_RAISED in self._event_handlers:
--> 464 self._fire_event(Events.EXCEPTION_RAISED, e)
465 else:
466 raise e
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/ignite/engine/engine.py:421, in Engine._fire_event(self, event_name, *event_args, **event_kwargs)
419 kwargs.update(event_kwargs)
420 first, others = ((args[0],), args[1:]) if (args and args[0] == self) else ((), args)
--> 421 func(*first, *(event_args + others), **kwargs)
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/scglue/models/base.py:163, in Trainer.fit.._handle_exception(engine, e)
161 engine.terminate()
162 else:
--> 163 raise e
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/ignite/engine/engine.py:840, in Engine._run_once_on_dataset(self)
838 self.state.iteration += 1
839 self._fire_event(Events.ITERATION_STARTED)
--> 840 self.state.output = self._process_function(self, self.state.batch)
841 self._fire_event(Events.ITERATION_COMPLETED)
843 if self.should_terminate or self.should_terminate_single_epoch:
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/scglue/models/scglue.py:728, in SCGLUETrainer.train_step(self, engine, data)
725 self.dsc_optim.step()
727 # Generator step
--> 728 losses = self.compute_losses(data, epoch)
729 self.net.zero_grad(set_to_none=True)
730 losses["gen_loss"].backward()
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/scglue/models/scglue.py:674, in SCGLUETrainer.compute_losses(self, data, epoch, dsc_only)
671 g_kl = D.kl_divergence(v, prior).sum(dim=1).mean() / vsamp.shape[0]
672 g_elbo = g_nll + self.lam_kl * g_kl
--> 674 x_nll = {
675 k: -net.u2x[k](
676 usamp[k], vsamp[getattr(net, f"{k}_idx")], xbch[k], l[k]
677 ).log_prob(x[k]).mean()
678 for k in net.keys
679 }
680 x_kl = {
681 k: D.kl_divergence(
682 u[k], prior
683 ).sum(dim=1).mean() / x[k].shape[1]
684 for k in net.keys
685 }
686 x_elbo = {
687 k: x_nll[k] + self.lam_kl * x_kl[k]
688 for k in net.keys
689 }
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/scglue/models/scglue.py:675, in (.0)
671 g_kl = D.kl_divergence(v, prior).sum(dim=1).mean() / vsamp.shape[0]
672 g_elbo = g_nll + self.lam_kl * g_kl
674 x_nll = {
--> 675 k: -net.u2x[k](
676 usamp[k], vsamp[getattr(net, f"{k}_idx")], xbch[k], l[k]
677 ).log_prob(x[k]).mean()
678 for k in net.keys
679 }
680 x_kl = {
681 k: D.kl_divergence(
682 u[k], prior
683 ).sum(dim=1).mean() / x[k].shape[1]
684 for k in net.keys
685 }
686 x_elbo = {
687 k: x_nll[k] + self.lam_kl * x_kl[k]
688 for k in net.keys
689 }
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/torch/distributions/negative_binomial.py:92, in NegativeBinomial.log_prob(self, value)
90 def log_prob(self, value):
91 if self._validate_args:
---> 92 self._validate_sample(value)
94 log_unnormalized_prob = (self.total_count * F.logsigmoid(-self.logits) +
95 value * F.logsigmoid(self.logits))
97 log_normalization = (-torch.lgamma(self.total_count + value) + torch.lgamma(1. + value) +
98 torch.lgamma(self.total_count))
File ~/miniconda3/envs/glue/lib/python3.9/site-packages/torch/distributions/distribution.py:288, in Distribution._validate_sample(self, value)
286 valid = support.check(value)
287 if not valid.all():
--> 288 raise ValueError(
289 "Expected value argument "
290 f"({type(value).name} of shape {tuple(value.shape)}) "
291 f"to be within the support ({repr(support)}) "
292 f"of the distribution {repr(self)}, "
293 f"but found invalid values:\n{value}"
294 )
ValueError: Expected value argument (Tensor of shape (128, 2000)) to be within the support (IntegerGreaterThan(lower_bound=0)) of the distribution NegativeBinomial(total_count: torch.Size([128, 2000]), logits: torch.Size([128, 2000])), but found invalid values:
tensor([[0.0000, 0.0000, 0.0000, ..., 0.0000, 0.0000, 0.0000],
[0.0000, 0.0000, 0.0000, ..., 0.0000, 0.0000, 0.0000],
[0.0000, 0.0000, 0.0000, ..., 0.0000, 0.0000, 0.0000],
...,
[0.0000, 0.0000, 0.0000, ..., 2.2001, 0.0000, 0.0000],
[1.0265, 0.0000, 0.0000, ..., 2.0998, 0.0000, 0.0000],
[0.0000, 0.0000, 0.0000, ..., 2.1896, 0.0000, 0.2278]],
device='cuda:0')
Hi Dr. Cao, sorry to disturb you. I met some problems when I run the tutorials:

I found that normalize_total will yield errors based on updated scanpy. I wonder if I use: normalize_per_cell is also ok or not. Thanks a lot.
Dear developers,
First thank you for providing such great package. And what I want to know is whether GLUE is able to integrate scATAC-seq and spatial RNA-seq data? If GLUE doesn't support spatial data, perhaps I can just treat them as typical scRNA-seq data, is it feasible? Thank you in advance!
Best,
Zhixin
Hello, I am Tianyu. Thanks for your great presentation in the single-cell competition workshop. After going through your code, I am a little confused about the method you used to generate the score matrix, I have two questions and I am looking forward to your reply.
Thanks a lot!

When I run case: Altas,i met the error in rule e02_cca_anchor:
e02_cca_anchor_nbconvert [4].log
How can I solve this problem?
Hi team! I see GLUE can successfully integrate multiple single-cell datasets cross modalities. I wonder if the cell types of one dataset are known, is it possible to transfer cell labels from one dataset to another? Just like what TransferData() do in Seurat. Many thanks!
In the docs you mention registering custom decoders/encoders which is exactly what I want to do. However, I'm not totally clear on how to achieve this since the functions referenced are internal. Is there a way to modify them in the API? Some python trick I am unaware of? Or is there some sort of register function for decoders/encoders like there is for datasets?
Hi, I was running into issues integrating my RNA and ATAC data and reproduced the same error even when using the same data from the tutorial. Here is what I am running:
glue = scglue.models.fit_SCGLUE(
{"rna": rna, "atac": atac}, graph,
fit_kws={"directory": "glue"}
)
returns this error.
Traceback (most recent call last):
File "<stdin>", line 3, in <module>
File "/home/klchiou/.local/lib/python3.7/site-packages/scglue/models/__init__.py", line 89, in fit_SCGLUE
pretrain = model(adatas, sorted(graph.nodes), **pretrain_init_kws)
File "/home/klchiou/.local/lib/python3.7/site-packages/scglue/models/scglue.py", line 1323, in __init__
u2c=None if all_ct.empty else sc.Classifier(latent_dim, all_ct.size)
File "/home/klchiou/.local/lib/python3.7/site-packages/scglue/models/base.py", line 273, in __init__
self._net = self.NET_TYPE(*args, **kwargs)
File "/home/klchiou/.local/lib/python3.7/site-packages/scglue/models/scglue.py", line 452, in __init__
super().__init__(g2v, v2g, x2u, u2x, idx, du, prior)
File "/home/klchiou/.local/lib/python3.7/site-packages/scglue/models/glue.py", line 232, in __init__
self.device = autodevice()
File "/home/klchiou/.local/lib/python3.7/site-packages/scglue/models/nn.py", line 178, in autodevice
best_devices = np.where(free_mems == free_mems.max())[0]
File "/home/klchiou/.local/lib/python3.7/site-packages/numpy/core/_methods.py", line 40, in _amax
return umr_maximum(a, axis, None, out, keepdims, initial, where)
ValueError: zero-size array to reduction operation maximum which has no identity
Help would be appreciated!
Hi Dr. Cao, sorry to disturb you again. When can we get access to CLUE working for the open problems competition? Thanks a lot.
Hi Dr. Cao, I found a strange thing in the tutorial. When I went to the training step, I met a bug:

I think that is caused by the codes in this part (preprocessing atac)

I cannot find the steps related to selecting hvgs of ATAC, and that is why I received the previous bugs.
Could you please help me address this problem? Thanks a lot!
Hi,
I followed the steps in the documentation to process my data, but an error occurred while running scglue.models.fit_SCGLUE. I'm not sure where the index numbers came from, or how to fix the mistake. Thanks.

Hi, is it possible to help upload the example data "Luo-2017.h5ad" and "10x-ATAC-Brain5k.h5ad" that you used in TripleOmics to pku ftp as well?
Thx a lot! Really amazing manuscript!
Hi, I'm going through the tutorial. The h5ad files used in the dataset cannot be downloaded, they require a password, e.g., ftp://ftp.cbi.pku.edu.cn/pub/glue-download/Chen-2019-RNA.h5ad.
I was wondering if you could point me to a downloadable h5ad file please.
Hi, thank you for your excellent tools!
Recently, our collaborator generated a series of 10X multiome datasets, and we hope to use GLUE to our paired datasets.
I found the class PairedSCGLUEModel in your "readthedocs" tutorial. Is there a tutorial for this class and applying GLUE to paired-datasets?
Thanks!
Hi, I have the error when running scglue.genomics.rna_anchored_prior_graph(rna, atac) when using the data provided in the tutorial. The error stated the bedtools version should be >=2.29.2, however, the bedtools is 2.30.0. I even reinstalled everything. Unfortuenly, the error didn't disappear. I would be appreciate if you can help me to figure it out. Thanks!


!
Hi
I am following evalution pipeline from the GLUE publication to reproduce the results ,and meet a error.
timeout 24h Rscript workflow/scripts/run_LIGER.R --input-rna results/raw/Chen-2019/subsample_size:8000-subsample_seed:2/rna.h5ad --input-atac results/raw/Chen-2019/subsample_size:8000-subsample_seed:2/frags2rna.h5ad -s 0 --output-rna results/raw/Chen-2019/subsample_size:8000-subsample_seed:2/null/LIGER_FiG/default/seed:0/rna_latent.csv --output-atac results/raw/Chen-2019/subsample_size:8000-subsample_seed:2/null/LIGER_FiG/default/seed:0/atac_latent.csv --run-info results/raw/Chen-2019/subsample_size:8000-subsample_seed:2/null/LIGER_FiG/default/seed:0/run_info.yaml > results/raw/Chen-2019/subsample_size:8000-subsample_seed:2/null/LIGER_FiG/default/seed:0/run_LIGER_FiG.log 2>&1 || touch results/raw/Chen-2019/subsample_size:8000-subsample_seed:2/null/LIGER_FiG/default/seed:0/.blacklist
Waiting at most 5 seconds for missing files.
MissingOutputException in line 120 of /root/GLUE/evaluation/workflow/rules/methods.smk:
Job Missing files after 5 seconds:
results/raw/Chen-2019/subsample_size:8000-subsample_seed:2/null/LIGER_FiG/default/seed:0/rna_latent.csv
results/raw/Chen-2019/subsample_size:8000-subsample_seed:2/null/LIGER_FiG/default/seed:0/atac_latent.csv
results/raw/Chen-2019/subsample_size:8000-subsample_seed:2/null/LIGER_FiG/default/seed:0/run_info.yaml
This might be due to filesystem latency. If that is the case, consider to increase the wait time with --latency-wait.
Job id: 28634 completed successfully, but some output files are missing. 28634
Job failed, going on with independent jobs.
Select jobs to execute...
Check the corresponding log file(results/raw/Chen-2019/subsample_size:8000-subsample_seed:2/null/LIGER_FiG/default/seed:0/run_LIGER_FiG.log):
Error in py_call_impl(callable, dots$args, dots$keywords) :
OSError: Unable to open file (file signature not found)
Can someone please explain the error?
I can only use the HPC cluster using PBS scheduling system, I should modify what files to be able to run Evalution?
高歌教授、曹志杰博士您好,十分感谢你们在15号晚上关于GLUE的研究分享,你们在里面提出了GLUE首次实现了百万级别单细胞数据的整合,那么使用GLUE进行百万级别单细胞数据的整合分析时,服务器需达到什么规模的计算资源呢(例如内存大小)?以及是否支持服务器集群的并行运算?能否分享一下你们在文中提出整合”人类胎儿全器官单细胞转录组和染色质开放组图谱“这个数据时所占用的服务器资源配置吗?
期待你们的回复,谢谢!
Hi,
I found some meta data files of the "data/download" folder are missed, such as for SNARE-seq (AdBrainCortex_SNAREseq_metadata.rds) and SHARE-seq (celltype.txt and celltype_v2.txt). Tough download links of the processed data can be found from data.rst, is it possible to provide the download link of these files for repeating. Thanks.
Hi,
Thank you for this great method! I have a question on specifically how to construct a guidance graph manually. In my setting, I directly create a data frame called reg2gene that is shown as:
'0' '1'
gene1 region1
gene2 region2
......
How can I convert this data frame into a graph that can be used in your method? I tried to use G = nx.from_pandas_edgelist(reg2gene, 0, 1), but the resulting graph doesn't seem able to be used in GLUE.
Yours sincerely,
Siqi Shen
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.
{kind=link}
{kind=link}